Enlace a Legislación Relacionada
 Sin Vigencia
Sin Vigencia
(APROBAR EL REGLAMENTO TÉCNICO CENTROAMERICANO RTCA 11.03.59:11 PRODUCTOS FARMACÉUTICOS, MEDICAMENTOS DE USO HUMANO, REQUISITOS DE REGISTRO SANITARIO, EN LA FORMA QUE APARECE COMO ANEXO 1 DE LA PRESENTE RESOLUCIÓN)
RESOLUCIÓN N°. 333-2013 (COMIECO-LXVI), aprobada el 12 de diciembre del 2013
Publicada en La Gaceta Diario Oficial N°. 173 del 11 de septiembre del 2014
EL CONSEJO DE MINISTROS DE INTEGRACIÓN ECONÓMICA
CONSIDERANDO:
Que de conformidad con los artículos 38, 39 y 55 del Protocolo al Tratado General de Integración Económica Centroamericana - Protocolo de Guatemala-, modificado por la Enmienda del 27 de febrero de 2002, el Consejo de Ministros de Integración Económica (COMIECO) tiene bajo su competencia los asuntos de la Integración Económica Centroamericana y, como tal, le corresponde aprobar los actos administrativos del Subsistema Económico;
Que de acuerdo con el artículo 15 de ese mismo instrumento jurídico regional, los Estados Parte tienen el compromiso de constituir una Unión Aduanera entre sus territorios, la que se alcanzará de manera gradual y progresiva, sobre la base de programas que se establezcan al efecto, aprobados por consenso;
Que en el marco del proceso de conformación de la Unión Aduanera, los Estados Parte han alcanzado importantes acuerdos en materia de Requisitos de Registro Sanitario de Medicamentos de Uso Humano, que requieren la aprobación del Consejo;
Que los Estados Parte, en su calidad de Miembros de la Organización Mundial del Comercio (OMC), notificaron al Comité de Obstáculos Técnicos al Comercio, de conformidad con lo establecido en el Acuerdo sobre Obstáculos Técnicos al Comercio, el Proyecto de Reglamento Técnico Centroamericano RTCA 11.03.59:11 Productos Farmacéuticos. Medicamentos de Uso Humano. Requisitos de Registro Sanitario; y el Procedimiento para el Reconocimiento Mutuo de Registros Sanitarios de Medicamentos de Uso Humano;
Que los Estados Parte, concedieron un plazo prudencial a los Estados Miembros de la OMC para hacer observaciones al proyecto de Reglamento notificado tal y como lo exige el numeral 4), párrafo 9 del artículo 2 del Acuerdo sobre Obstáculos Técnicos al Comercio, observaciones que fueron debidamente analizadas y atendidas en lo pertinente;
Que de conformidad con el Acuerdo sobre Obstáculos Técnicos al Comercio, los Miembros preverán un plazo prudencial entre la aprobación de los reglamentos técnicos y su entrada en vigor, con el fin de dar tiempo a los productores para adaptar sus productos o sus métodos de producción a lo establecido en los reglamentos;
Que de conformidad con el párrafo 3 del Artículo 55 del Protocolo de Guatemala, se recabó la opinión del Comité Consultivo de Integración Económica,
POR TANTO:
Con fundamento en lo dispuesto en los artículos 1, 3, 5, 7, 15, 26, 30, 36, 37, 38, 39, 46, 52 y 55 del Protocolo al Tratado General de Integración Económica Centroamericana -Protocolo de Guatemala-,
RESUELVE:
1. Aprobar el Reglamento Técnico Centroamericano RTCA 11.03.59: 11 PRODUCTOS FARMACÉUTICOS. MEDICAMENTOS DE USO HUMANO. REQUISITOS DE REGISTRO SANITARIO, en la forma que aparece como Anexo 1 de la presente Resolución y que forma parte integrante de la misma.
2. En el caso de medicamentos que se encuentren dentro de las categorías mencionadas en el numeral 7. 11.3 que se vayan a renovar y que originalmente su seguridad y eficacia no hayan sido documentados según los requisitos sanitarios descritos en el presente reglamento, deberán presentar los requisitos establecidos en dicho numeral.
3. En el caso de El Salvador la renovación del registro realizado anteriormente a la vigencia de este RTCA para las categorías de medicamentos contempladas en el numeral 7. 11.3 se excluye de lo establecido en este Reglamento.
4. Aprobar el Procedimiento para el Reconocimiento Mutuo de Registro Sanitario de Medicamentos de Uso Humano, tal como aparece Anexo 2 a la presente Resolución.
5. Este procedimiento no aplica para las asociaciones de medicamentos registradas en El Salvador previamente a la fecha de entrada en vigor del RTCA 11.03.59: 11 Productos Farmacéuticos. Medicamentos de Uso Humano. Requisitos de Registro Sanitario.
6. La presente Resolución entrará en vigencia el 12 de junio de 2014 y será publicada por los Estados Parte.
7. No obstante lo establecido en el numeral anterior, la presente Resolución no entrará en vigor para Panamá, hasta que este Consejo emita el acto administrativo correspondiente, conforme a lo establecido en el Artículo Transitorio del Protocolo de Incorporación de la República de Panamá al Subsistema de Integración Económica.
Panamá, República de Panamá, 12 de diciembre de 2013.
(f) Anabel González Campabadal, Ministra de Comercio Exterior de Costa Rica. (f) Mario Roger Hernández, Viceministro, en representación del Ministro de Economía de El Salvador. (f) Sergio de la Torre Gimeno, Ministro de Economía de Guatemala. (O José Adonis Lavaire, Ministro de Industria y Comercio de Honduras. (f) Orlando Solórzano Delgadillo, Ministro de Fomento, Industria y Comercio de Nicaragua. (f) Ricardo A. Quijano Jiménez, Ministro de Comercio e Industrias de Panamá.ANEXO 1 DE LA RESOLUCIÓN No. 333-2013
(COMIECO-LXVI)
REGLAMENTO RTCA 1 1.03.59:11
TÉCNICO CENTROAMERICANO
PRODUCTOS FARMACÉUTICOS. MEDICAMENTOS PARA USO HUMANO. REQUISITOS DE REGISTRO SANITARIO.
CORRESPONDENCIA: Este Reglamento no tiene correspondencia con ninguna otra norma o reglamento internacional.
ICS 11.120.01 RTCA 11.03.59:11
Reglamento Técnico Centroamericano, editado por:
· Ministerio de Economía y Comercio, MINECO
· Organismo Salvadoreńo de Reglamentación Técnica, OSARTEC
· Ministerio de Fomento, Industria y Comercio, MIFIC
· Secretaría de Industria y Comercio, SIC
· Ministerio de Economía, Industria y Comercio, MEIC
INFORME
Los respectivos Comités Técnicos de Reglamentación Técnica de los países centroamericanos y sus sucesores, son los organismos encargados de realizar el estudio o la adopción de los Reglamentos Técnicos. Están conformados por representantes de los sectores Académico, Consumidor, Empresa Privada y Gobierno.
Este documento fue aprobado como Reglamento Técnico Centroamericano, RTCA 11.03.59: 11 Productos Farmacéuticos. Medicamentos para uso Humano. Requisitos de Registro Sanitario, por el Sub grupo de Medicamentos y Productos Afines y el Subgrupo de Medidas de Normalización. La oficialización de este reglamento técnico, conlleva la ratificación por el Consejo de Ministros de Integración Económica Centroamericana (COMIECO).
MIEMBROS PARTICIPANTES
Por Guatemala
Ministerio de Salud Pública y Asistencia Social
Por El Salvador
Dirección Nacional de Medicamentos
Por Honduras
Secretaria de Salud
Por Costa Rica
Ministerio de Salud
Por Nicaragua
Ministerio de Salud
1. OBJETO
Establecer las condiciones y requisitos bajo las cuales se otorgará el registro sanitario de los medicamentos para uso humano.
2. CAMPO DE APLICACIÓN
Aplica a los medicamentos para uso humano que fabrican o importan personas naturales o jurídicas para su comercialización en el territorio centroamericano. Se excluyen del presente Reglamento las preparaciones magistrales.
NOTAS:
1. Los medicamentos biológicos y biotecnológicos serán registrados de conformidad con la legislación nacional de cada Estado Parte.
2. Mientras un Estado Parte no cuente con una legislación nacional para el registro de medicamentos biológicos y biotecnológicos, a éstos productos se les aplicará el presente reglamento.
3. En el caso de los suplementos nutricionales o alimenticios en tanto no exista un reglamento armonizado a nivel regional, se aplicará la legislación de cada estado parte.
3. DOCUMENTOS A CONSULTAR
3.1 RTCA 11.03.39:06 Productos Farmacéuticos. Reglamento de Validación de Métodos Analíticos para la Evaluación de la Calidad de los Medicamentos Vigente.
3.2 RTCA 11.01.02:04 Productos Farmacéuticos. Etiquetado de Productos Farmacéuticos para Uso Humano Vigente.
3.3 RTCA 11.01.04:10 Productos Farmacéuticos. Estudios de Estabilidad de Medicamentos para Uso Humano Vigente.
3.4 RTCA 11.03.56:09 Productos Farmacéuticos. Medicamentos para Uso Humano. Verificación de la Calidad Vigente.
3.5 RTCA Productos Farmacéuticos. Medicamentos de Uso Humano. Buenas Prácticas de Manufactura para la Industria Farmacéutica Vigente.
4. DEFINICIONES
4.1 Acondicionamiento: todas las operaciones, incluidas el llenado y el etiquetado, necesarias para convertir un producto a granel en un producto terminado.
4.2 Autoridad reguladora: autoridad responsable de la regulación sanitaria en cada país o región.
4.3 Autoridad reguladora de referencia regional: es la autoridad reguladora nacional competente y eficiente en el desempeńo de las funciones de regulación sanitaria recomendadas por la OPS/ OMS, para garantizar la calidad, inocuidad y eficacia de los medicamentos y productos biológicos.
4.4 Autoridad reguladora estricta: son aquellas definidas en el proceso de precalificación de productos farmacéuticos de la OMS.
4.5 Buenas Prácticas de Manufactura: conjunto de procedimientos y normas destinados a garantizar la producción uniforme de los lotes de productos farmacéuticos que cumplan las normas de calidad.
4.6 Certificado de libre venta: documento expedido por la Autoridad Reguladora del país o región de origen, o procedencia, en donde se certifica que el medicamento a que se refiere el certificado tiene su registro vigente y está autorizado para la venta en ese país; en el caso de fabricación por terceros o filiales y que el producto no se comercialice en el país de origen, podrá ser expedido por la autoridad reguladora del país del titular.
4. 7 Certificado de producto farmacéutico: certificación propuesta por la OMS y emitida por la Autoridad Reguladora del país o región de origen o procedencia, como parte del sistema de certificación de la calidad de los productos farmacéuticos objeto de comercio internacional, en el caso de fabricación por terceros o filiales y que el producto no se comercialice en el país de origen, podrá ser expedido por la Autoridad Reguladora del país del titular.
4.8 Certificado de Buenas Prácticas de Manufactura: documento expedido por la Autoridad Reguladora del país en el cual se encuentra ubicado el laboratorio fabricante donde se certifica que el laboratorio cumple con las Buenas Prácticas de Manufactura.
4.9 Co-empaque: forma de presentación de dos o más productos previamente registrados que se comercializan en forma conjunta para el tratamiento de una patología específica.
4.10 Contrato de fabricación: documento legal celebrado entre el titular del medicamento y el fabricante en el cual se establecen las condiciones, compromisos y demás circunstancias para la fabricación de uno o más productos.
4.11 Denominación común internacional: nombre recomendado por la OMS para los ingredientes activos de los medicamentos.
4.12 Documento oficial: aquel emitido por la Autoridad Competente del Estado.
4.13 Etiquetado o rotulado: es toda inscripción o leyenda que identifica al producto, que se imprima, adhiera o grabe en el envase o empaque primario, y/o envase o empaque secundario.
4.14 Envase primario o empaque primario: recipiente dentro del cual se coloca directamente el medicamento en la forma farmacéutica terminada.
4.15 Envase secundario o empaque secundario: recipiente dentro del cual se coloca el envase primario que contiene al medicamento en su forma farmacéutica terminada para su distribución y comercialización.
4.16 Entidad química: grupo funcional del princ1p10 activo que es responsable de la acción fisiológica o farmacológica. Se entiende que comparten una entidad química todos aquellos polimorfos, isómeros y aquellos derivados con partes unidas a la entidad química que constituyen un Ester, éter, sal, (incluyendo una sal con uniones de hidrógeno o coordinadas), u otro derivado no covalente tales como los complejos entre otros.
4.17 Equivalente farmacéutico: medicamento que contiene idénticas cantidades de los mismos principios activos del producto al que es equivalente, la misma sal o éster del principio activo, en idénticas formas farmacéuticas, pero que puede o no contener los mismos excipientes. En consecuencia, dos equivalentes farmacéuticos pueden mostrar diferentes biodisponibilidades, magnitudes y perfiles temporales de sus actividades farmacológicas.
4.18 Fabricación o manufactura: todas las operaciones involucradas en la compra de materiales y productos, producción, acondicionamiento, control de calidad, aprobación, almacenamiento, distribución del producto terminado y los controles relacionados.
4.19 Fabricación por terceros: fabricación nacional o extranjera realizada dentro de los límites de una contratación previa entre el titular del medicamento y el fabricante, siendo el titular el responsable del producto.
4.20 Inserto, prospecto o instructivo: información técnico científica que se adjunta al producto terminado, el cual debe contener los datos necesarios para el uso seguro y eficaz del medicamento que lo contiene.
4.21 Laboratorio fabricante: entidad autorizada con instalaciones diseńadas, para realizar todas las operaciones que involucran la fabricación de productos farmacéuticos.
4.22 Medicamento multiorigen: producto que es equivalente farmacéutico y que puede o no ser equivalente terapéutico. Los medicamentos multiorigen que son equivalentes terapéuticos son intercambiables.
4.23 Medicamento huérfano: el que se destina al tratamiento de una enfermedad rara, grave o que produzca incapacidad y cuyo interés comercial resulta poco probable, o sin medidas de estímulo. Van destinados a un reducido grupo de pacientes pero responde a necesidades de salud pública.
4.24 Modificaciones post-registro: modificaciones al registro sanitario de un producto farmacéutico posterior al otorgamiento de su registro.
4.25 Monografía del producto: descripción científico - técnica del perfil de seguridad y eficacia de un medicamento o producto farmacéutico.
4.26 País de origen: país donde se fabrica el producto. En caso que en la fabricación intervenga más de un laboratorio fabricante, el país de origen es aquel en que se realiza la fabricación de al menos el producto a granel.
4.27 País de procedencia: país desde donde se distribuye, acondiciona o exporta el producto. Siempre que estos intervengan en el proceso de fabricación; al menos hasta el empaque primario.
4.28 País del titular: país donde está domiciliado el titular del producto.
4.29 Preparaciones magistrales: producto medicinal elaborado por el farmacéutico en una farmacia para atender una prescripción o receta médica de un paciente individual.
4.30 Principio activo: sustancia dotada de un efecto farmacológico específico o que sin poseer actividad, al ser administrado en el organismo la adquiere luego que sufren cambios en su estructura química.
4.31 Producto farmacéutico o medicamento: sustancia simple o compuesta, natural, sintética, o mezcla de ellas, con forma farmacéutica definida, empleada para diagnosticar, tratar, prevenir enfermedades o modificar una función fisiológica de los seres humanos.
4.32 Producto a granel: producto que ha pasado por todas las fases de producción excepto el empaque primario.
4.33 Producto terminado: el que está en su envase definitivo, rotulado y listo para ser distribuido y comercializado.
4.34 Profesional responsable: profesional farmacéutico o químico farmacéutico responsable del trámite de registro sanitario ante la Autoridad Reguladora, autorizado por el titular del medicamento o su representante legal a través de un poder otorgado de acuerdo a la legislación de cada Estado parte.
4.35 Registro sanitario: aprobación por la autoridad reguladora de un país para la comercialización de un medicamento, una vez que el mismo ha pasado el proceso de evaluación relativo a la calidad, eficacia y seguridad.
4.36 Representante legal: persona natural o jurídica que reside en el país donde se tramita el registro, autorizada por el titular del medicamento, a través de un poder otorgado de acuerdo a la legislación de cada Estado parte, para que responda ante la Autoridad Reguladora.
4.37 Sistema envase-cierre: conjunto de materiales de empaque que contienen y protegen la forma farmacéutica, incluye tanto el empaque primario, como el secundario si este último tiene la función de proporcionar protección adicional al producto.
4.38 Titular del producto o titular del registro: persona natural o jurídica propietaria del producto.
5. SÍMBOLOS Y ABREVIATURAS
5.1. OMS: Organización Mundial de la Salud.
5.2. FDA: Administración de Drogas y Alimentos de los Estados Unidos de América.
5.3. ATC: Clasificación Anatómica y Terapéutica.
5.4. EMA: Agencia Europea de Medicamentos.
5.5. BPM: Buenas Prácticas de Manufactura.
5.6. DCI: Denominación Común Internacional.
5.7. USPDI: Drug Information for the Health Care Professionals.
5.8. AOAC: Association of Analytical Chemists.
6. DISPOSICIONES GENERALES
6.1 Para la importación, distribución, comercialización, prescripción y promoción, todo medicamento requiere previamente su registro sanitario ante la Autoridad Reguladora.
6.2 El registro sanitario de medicamentos tendrá una vigencia de cinco ańos, reservándose la Autoridad Reguladora el derecho de suspender o cancelar el mismo cuando hayan razones sanitarias de carácter científico técnico o legales debidamente justificadas.
6.3 Todo certificado o documento oficial requerido debe de estar vigente al momento de su presentación. Los documentos oficiales tendrán la validez que les otorgue la Autoridad Reguladora del país donde se emite. En los casos en los que no se indique la vigencia, ésta será de 2 ańos para efecto del trámite de registro, a partir de la fecha de emisión.
6.4 Todo documento oficial o legal debe presentarse en original copia legalizada de conformidad a la legislación de cada Estado parte.
El documento se deberá presentar en idioma espańol/castellano o acompańado de su respectiva traducción emitida de conformidad con la legislación de cada Estado parte.
6.5 No se permiten correcciones en las certificaciones o en los documentos oficiales presentados, a menos que estén sustentadas por la misma instancia que emitió el documento original.
6.6 Todo documento oficial o legal emitido en el extranjero debe legalizarse cumpliendo con la normativa nacional específica.
6.7 En aquellos casos en que aplique y para efectos del registro de un medicamento específico, se permitirá que el solicitante haga referencia a documentos originales, vigentes que consten en archivos de la Autoridad Reguladora. En este caso el solicitante debe hacer referencia de la gestión en la cual se entregó el documento original, presentando fotocopia simple del mismo.
6.8 En los casos de productos a registrar que no se comercialicen en el país de origen o procedencia la Autoridad Reguladora evaluará la justificación aportada.
6.9 Corresponden a un mismo registro:
6.9.1 Diferentes presentaciones comerciales de medicamentos con la misma concentración y la misma forma farmacéutica.
6.9.2 Medicamentos con igual formula cuali – cuantitativa y diferente sabor y/o color.
6.10 Libros Oficiales.
Para efecto del presente reglamento los libros reconocidos como oficiales son:
6.10.1 Para verificación de la calidad:
Para el registro de medicamentos cuyos métodos de análisis para producto terminado sean farmacopeicos, éstos deben estar descritos en la versión que incluya pruebas y especificaciones recientes que permitan evaluar la calidad del medicamento para la forma farmacéutica específica. En caso de no utilizar la versión más actualizada, el solicitante deberá justificar esa omisión, declarando cuál es la farmacopea y edición que utiliza, siempre y cuando cumpla con lo establecido en el Reglamento Técnico Centroamericano de Verificación de la Calidad vigente y con el listado de libros oficiales. La Autoridad Reguladora valorará la justificación aportada. En caso de medicamentos descritos en más de una farmacopea, se debe utilizar la que indique las especificaciones más actuales y completas para verificar la calidad de los productos.
Los libros oficiales son los siguientes:
a) Farmacopea Alemana
b) Farmacopea Argentina
c) Farmacopea Británica
d) Farmacopea de los Estados Unidos (USP) y el Formulario Nacional de los Estados Unidos (USP/NF)
e) Farmacopea Espańola
f) Farmacopea Europea
g) Farmacopea Francesa
h) Farmacopea Helvética
i) Farmacopea Internacional
j) Farmacopea Japonesa
k) Farmacopea Mexicana
l) Farmacopea China
m) Food Chemical Codex (FCC)
n) AOAC
o) otras que en consenso los países acuerden incluir.
6.10.2 Para evaluación farmacológica:
Se podrá utilizar la siguiente literatura para la evaluación de la Monografía:
a. Drug Information for the Health Care Professionals (USPDJ)
b. Drug Information (AHFS).
c. Martindale. The Extra Pharmacopoeia.
d. Normas Farmacológicas de Centroamérica y República Dominicana.
e. Libros de farmacología que estén fundamentados científicamente.
f. Artículos completos de revistas fundamentados científicamente.
6.11. El nombre del medicamento a registrar no debe causar confusión con otro ya registrado, ya sea en su forma escrita o pronunciada, por ello la denominación del medicamento a registrar debe cumplir las siguientes condiciones:
6.11.1 No se permite el registro de medicamentos con un mismo nombre de marca comercial y diferentes principios activos, ni se acepta la utilización de un nombre de marca comercial que haya sido usado anteriormente para productos de diferente indicación.
6.11.2 Sólo se acepta el registro usando el mismo nombre de marca comercial, en el caso de medicamentos declarados de venta libre que sean utilizados con indicaciones terapéuticas similares aunque contengan principios activos diferentes (líneas de tratamiento).
6.11.3 Se acepta el registro de productos poli fármacos de venta libre utilizando como parte del nombre una acción terapéutica.
6.11.4 Se acepta en el nombre de un medicamento el uso del nombre del titular o sus siglas acompańado de la denominación común internacional de los principios activos.
6.11.5 El nombre del medicamento, los logos u otras frases no debe tener connotaciones terapéuticas que puedan generar confusión en las indicaciones de uso.
6.11.6 El nombre del medicamento a registrar debe coincidir con toda la documentación presentada, en caso contrario se debe presentar una nota aclaratoria firmada por el titular o su representante legal debidamente legalizada si está autorizado para ello, en donde especifique que todos los documentos corresponden al mismo medicamento.
6.11. 7 Un medicamento puede designarse con un nombre de marca comercial o bien con una denominación común internacional. Cuando sea una marca comercial no puede confundirse con una denominación común internacional ni que induzca a error sobre las propiedades terapéuticas o la naturaleza del medicamento.
6.12. Medicamentos co-empacados.
6.12.1 En caso de medicamentos co-empacados se debe registrar cada medicamento por separado y posteriormente solicitar una modificación del registro para el co-empaque.
En caso que algunos de los productos no esté registrado deberá registrarse y posteriormente solicitar el co-empaque. La fecha de vencimiento otorgada será la del registro que se vence primero.
6.12.2 En caso se solicite la comercialización de varios productos para un tratamiento específico en un solo empaque y los productos no estén registrados previamente, debe iniciar el trámite de registro, cumpliendo con los requisitos de cada producto incluidos en el empaque.
6.13. Para el etiquetado de los medicamentos a los que se refiere este reglamento deberá regirse lo indicado en el Reglamento Técnico Centroamericano de Etiquetado de Productos Farmacéuticos para uso Humano vigente.
Toda información presente en la etiqueta o impresa en el empaque del medicamento o inserto, prospecto o instructivo dirigido a los profesionales o pacientes, deberá apegarse a la información aprobada en el registro.
6.14. Si un mismo producto es fabricado en diferentes filiales o países, deberá de tramitar un Registro Sanitario por cada país o laboratorio fabricante.
NOTA: En el caso de El Salvador no aplica esta disposición, ya que en su procedimiento registral se establece la fabricación alterna.
6.15. Para garantizar la calidad de los medicamentos, las Autoridades Reguladoras podrán verificar el cumplimiento de las Buenas Prácticas de Manufactura por los medios que consideren necesarios, incluyendo la inspección en las instalaciones de los laboratorios fabricantes establecidos dentro y fuera de los países Centroamericanos, aplicando el Reglamento Técnico Centroamericano de Productos Farmacéuticos Medicamentos de Uso Humano Buenas Prácticas de Manufactura para la Industria Farmacéutica vigente.
La Autoridad Reguladora podrá solicitar a las autoridades reguladoras de referencia regional y autoridades reguladoras estrictas acreditadas por la OMS, la verificación del cumplimiento de Buenas Prácticas de Manufactura de laboratorios farmacéuticos que ellos hayan inspeccionado.
6.16. En materia de propiedad intelectual, se aplicará la normativa vigente de cada país.
6.17. Estudios de equivalencia terapéutica se aplicará la normativa vigente de cada país.
6.18. El procedimiento administrativo para el trámite de registro sanitario, renovación y modificaciones se hará de acuerdo a la legislación interna de cada Estado Parte.
6.19. El incumplimiento del presente reglamento dará lugar a la aplicación de lo establecido en el régimen sancionatorio de cada estado parte.
7. REQUISITOS PARA REGISTRO SANITARIO
Los requisitos para registro sanitario son los siguientes:
7.1. Solicitud firmada y sellada por el Profesional Responsable conteniendo la información detallada en el Anexo 2 de este Reglamento.
7.2. Poderes que acrediten la representación legal y/o técnica otorgada por el titular a la(s) persona(s) natural (les) o jurídica(s), de acuerdo a la legislación de cada país, (original o fotocopia autenticada del documento).
7.3. Certificado de producto farmacéutico tipo OMS, el cual deberá presentarse en original o fotocopia autenticada del documento legalizado.
En caso que no se emita este tipo de certificado, se admite la presentación de:
7.3.1. Certificado de Libre Venta En caso de presentar un certificado que avale dos o más productos se aceptará fotocopia autenticada y/o certificada del documento legalizado.
7.3.2. Certificado de Buenas Prácticas de Manufactura de cada uno de los establecimientos que intervienen en la fabricación del producto, para la forma farmacéutica y tipo de producto específico a registrar, extendido por la Autoridad Reguladora del país o países en donde se lleva a cabo el proceso de fabricación, original legalizado o fotocopia autenticada del mismo, éste debe indicar que cumple con la normativa de buenas prácticas de manufactura.
7.4. Contrato de fabricación o en su defecto el extracto relativo a las partes del contrato de fabricación, cuando aplique, en original o fotocopia autenticada o certificada del documento legalizado, que contenga al menos la siguiente información:
a) Firmado por el titular y el fabricante en forma conjunta o por separado.
b) Compromiso de cumplimiento de las Buenas Prácticas de Manufactura.
c) Establecer las condiciones de producción, análisis cuando aplique, o cualquier otra gestión técnica relacionada con estos.
d) Debe describir el manejo de materias primas, material de acondicionamiento, material a granel y producto terminado y en el caso que sean rechazados.
e) Permitir el ingreso del contratante a las instalaciones del contratista (contratado) para auditorias.
f) Permitir el ingreso del contratista (contratado) a las instalaciones del contratante.
g) Listar cada uno de los productos o servicios de análisis objeto del contrato.
7.5. Fórmula cuantitativa y cualitativa completa del producto por unidad de dosis. Se debe presentar en original firmada y sellada por el profesional responsable del laboratorio fabricante o titular del producto.
Además declarar lo siguiente:
7.5.1. Todos los componentes del medicamento deben ser descritos con su denominación común o genérica internacionalmente aceptada y no deben presentarse con siglas ni abreviaciones, las unidades deben estar dadas según el Sistema Internacional (SI). En caso de principios activos en forma de sales, ésteres u otros, se deberá declarar la cantidad equivalente de la molécula a la que se refiere la dosis terapéutica.
7.5.2. Composición del sistema de liberación para los productos de liberación modificada.
7.5.3. Composición cualitativa de las cápsulas vacías.
7 .5.4. Composición cualitativa de las tintas de impresión en las cápsulas, grageas y tabletas recubiertas.
7 .5.5. Declaración cualitativa de los disolventes orgánicos clase 2 ó 3, utilizados en el proceso de fabricación.
No se permitirá el registro de medicamentos que utilicen disolventes orgánicos Clase 1.
NOTA: Los disolventes orgánicos mencionados en el párrafo anterior son los establecidos en USP a partir de la edición 31.
7.5.6. Los excesos de principios activos utilizados en la fabricación.
7.5.7. En el caso de cremas y ungüentos, la concentración debe expresarse por cada gramo, 100g o en porcentaje. Las lociones, colirios, sesiones tópicas e infusiones parenterales deben expresar su concentración por mL, 100 mL o en porcentaje.
Dicha fórmula completa podrá estar contemplada en el Certificado de Libre Venta o en el Certificado del Producto Farmacéutico tipo OMS, lo que eximirá de presentarlo en forma individual.
7.6. Monografía del producto.
La información incluida dentro de la Monografía deberá estar fundamentada en los libros oficiales. En caso de divergencias con tales libros o si el medicamento no está descrito en ellos, se deberá presentar la información científica que lo respalde, la cual será evaluada por la Autoridad Reguladora.
Toda monografía debe corresponder a la forma farmacéutica del medicamento a registrar, sin embargo, podrá incluir otras presentaciones o concentraciones siempre que se incluya la que se está registrando. La cual debe contener la siguiente información:
a) Denominación común o genérica internacionalmente aceptada y concentración del medicamento.
b) Forma farmacéutica.
c) Estructura, nombre químico del principio activo o en su defecto adjuntar la ficha técnica que declare esta información.
d) Farmacología clínica.
e) Indicaciones.
f) Contraindicaciones.
g) Precauciones y advertencias.
h) Interacciones.
i) Efectos adversos.
j) Dosis y administración.
k) Recomendación en caso de sobredosificación según el perfil toxicológico.
l) Abuso y adicción.
m) Fecha de revisión de la monografía.
n) Lista de referencias bibliográficas completas.
o) Categoría terapéutica según Clasificación Anatómica Terapéutica (ATC), en el subgrupo farmacológico versión actualizada.
p) Forma de preparación
NOTA: Cuando la información solicitada no sea aplicable a las características propias del producto, podrá obviarse en la monografía.
7.7. Métodos de análisis validados según el Reglamento Técnico Centroamericano de Validación de Métodos Analíticos para la evaluación de la calidad de los medicamentos vigente, adjuntando el informe del estudio de validación correspondiente.
7.8 Especificaciones organolépticas, físicas, químicas, biológicas y microbiológicas del producto terminado que cumplan con lo establecido en el Reglamento Técnico Centroamericano de Verificación de la Calidad vigente. Los medicamentos con concentraciones mayores del 30% de alcohol; así como los que por su naturaleza son antisépticos, quedan exentos de presentar especificaciones microbiológicas.
7.9. Etiquetas del envase/empaque primario, secundario e inserto en original o sus proyectos, conforme al Reglamento Técnico Centroamericano de Etiquetado de Productos Farmacéuticos para Uso Humano vigente.
7.10. Informe del Estudio de Estabilidad conforme al Reglamento Técnico Centroamericano de Estudios de Estabilidad de Medicamentos para uso Humano vigente.
7.11. Estudios de seguridad y eficacia. Todos los informes de los estudios clínicos deben haber sido elaborados en un periodo no mayor a 10 ańos o presentar su debida justificación si fuera mayor de este periodo. Los informes deben referirse al mismo medicamento que se presenta para su registro sanitario, en los siguientes casos:
7.11.1. Para medicamentos cuya seguridad y eficacia no ha sido documentada en la literatura oficial deberán presentar:
a) Informes concluyentes de los resultados de los estudios pre clínicos.
b) Informes concluyentes de los resultados de los estudios clínicos fase I, II y III.
NOTA: Estos estudios se aceptarán en formato electrónico, siempre y cuando la Autoridad Reguladora tenga acceso libremente a la información.
7.11.2. En caso de medicamentos con entidades químicas previamente registradas cuyo principio activo corresponde a nuevos polimorfos, isómeros y aquellos derivados con partes unidas a la entidad química que la constituyen como Ester, éter, sal (incluyendo una sal con uniones de hidrógeno o coordinadas), u otro derivado no covalente, tales como los complejos, entre otros, y no estén descritos en la literatura oficial deberán presentar Informes concluyentes de los resultados de los estudios clínicos fase I, II y III.
7.11.3. Para medicamentos que contienen princ1p1os activos previamente registrados pero que presentan una o más de las siguientes características:
a) Nuevas combinaciones fijas de principios activos.
b) Nueva forma farmacéutica con una vía de administración ya registrada.
c) Nueva forma farmacéutica con una nueva vía de administración.
d) Nueva forma farmacéutica con una nueva forma de liberación.
e) Nuevas potencias o concentraciones de principios activos previamente registrados.
f) Nuevas potencias o concentraciones de princ1p1os activos registrados con la misma vía de administración, forma farmacéutica y posología.
g) Nueva forma de liberación con la misma vía de administración de un medicamento previamente registrado.
h) Nuevas vías de administración con una forma farmacéutica ya registrada.
Se debe presentar los informes concluyentes de los estudios clínicos que comprueben el objetivo u objetivos planteados según las variaciones anteriormente citadas para el producto en evaluación y que demuestren a la Autoridad su calidad, seguridad y eficacia.
7.11.4 Si al evaluar la documentación se comprueba que la información presentada no es concluyente, la Autoridad Reguladora podrá solicitar estudios clínicos complementarios. En caso de que científicamente no proceda la presentación de algún estudio clínico complementario, el solicitante deberá presentar la justificación para su valoración por parte de la Autoridad Reguladora.
7.11.5 Una solicitud para la cual no se presente información sobre seguridad y eficacia debe ser considerada por los Estados parte como una solicitud para el registro de un producto farmacéutico multiorigen. Las Autoridades Reguladoras podrán otorgar el registro sanitario a un producto farmacéutico multiorigen que:
a) Sea un equivalente farmacéutico de un producto que tenga las siguientes características:
-Que la Autoridad Reguladora cuente con los datos sobre seguridad y eficacia.
-Se haya otorgado previamente un registro sanitario en el Estado parte a registrar y
-Que no se encuentre protegido por patentes o datos de prueba;
O
b) Sea un producto farmacéutico que cumpla las siguientes condiciones:
- Que el producto innovador no haya sido registrado en el país y que la autoridad sanitaria considere como una excepción o en caso de necesidades médicas.
- Que el solicitante presente un documento emitido o publicado por una autoridad reguladora de cualquier país, que demuestre que existe un producto innovador que ha sido autorizado para su comercialización en ese país. Este documento deberá demostrar una relación riesgo beneficio favorable del producto a registrar. La autoridad reguladora del Estado Parte podrá exigir que el producto innovador cuente con un plazo mínimo de comercialización que demuestre la relación riesgo beneficio favorable del producto a registrar. En relación con la protección de datos de prueba se aplicará la legislación nacional específica establecida para estos efectos.
-Que exista información internacionalmente reconocida (publicada en libros oficiales o por autoridades reguladoras estrictas o de referencia), que garantice la seguridad y eficacia del producto farmacéutico a registrar.
7.12. a) Estándares primarios o materias primas estandarizadas.
b) Estándares de las sustancias relacionadas y/o de los productos de degradación, cuando la metodología lo requiera.
En ambos casos con su respectiva trazabilidad mediante una fotocopia del certificado de análisis, a excepción de muestras de farmacopea oficial' que no cuentan con estos certificados.
La Autoridad Reguladora evaluará de acuerdo a riesgo sanitario, el requerimiento de presentar los estándares de las sustancias relacionadas y/o productos de degradación.
7.13. Muestras de producto terminado, según cantidad armonizada para realizar los análisis, de acuerdo al Reglamento Técnico Centroamericano de Verificación de la Calidad de los Medicamentos vigente.
Cuando el laboratorio oficial no cuente con la tecnología o la capacidad instalada para realizar los análisis de determinados medicamentos, la Autoridad Reguladora tendrá la potestad de solicitarlos a laboratorios externos nacionales o extranjeros que posean dicha tecnología, acreditados por las entidades competentes, respetando la cantidad de muestras que el laboratorio tenga establecido. Los costos de envío y análisis de las muestras serán pagados por el fabricante o importador, el resultado de los análisis será reconocido en los estados parte.
NOTA: En el caso de Costa Rica los requisitos 7.12 y 7.13 se solicitarán posterior al registro del medicamento.
7.14. Un ejemplar de producto terminado.
En aquellos medicamentos clasificados internacionalmente como radiactivos o armas biológicas la Autoridad Reguladora podrá eximir de la presentación de este requisito. Para los medicamentos que requieren cadena de frío, así como los citotóxicos y biológicos, se acepta presentar los envases sin el producto con el sistema de cierre con el que se van a comercializar.
NOTA: En el caso de Guatemala y El Salvador, no se solicitará el requisito ya que se utilizan las muestras de análisis.
En el caso de Costa Rica no se solicitará el requisito para el proceso de registro.
7.15. Comprobante de pago
8 REQUISITOS PARA PRODUCTOS CO-EMPACADOS
8.1 Comprobante de pago
8.2 Solicitud firmada y sellada por el Profesional Responsable, conteniendo la información detallada en el Anexo 2 de este Reglamento.
8.3 Proyecto de etiquetas del co-empaque y su inserto, conforme al Reglamento Técnico Centroamericano de Etiquetado de Productos Farmacéuticos para uso Humano vigente.
8.4 Información científica que respalde el esquema de tratamiento.
9 REQUISITOS PARA LA RENOVACIÓN DEL REGISTRO SANITARIO
La renovación del registro de un medicamento, podrá gestionarse al menos 3 meses antes de su vencimiento.
Una vez vencido el registro sanitario, no se aceptará la solicitud de renovación debiendo tramitarse como registro nuevo.
Si durante los 6 meses posteriores al vencimiento del registro de un medicamento el interesado solicita se le mantenga el número asignado presentando la causa justificada, la Autoridad Reguladora le mantendrá el número original, sin embargo, durante este periodo, no podrá comercializarlo.
No se podrá otorgar la renovación, hasta haber aprobado los cambios post registros solicitados.
En el caso de medicamentos que se encuentren dentro de las categorías mencionadas en el numeral 7. 11.3 que se vayan a renovar y que originalmente su seguridad y eficacia no hayan sido documentados según los requisitos sanitarios descritos en el presente reglamento, deberán presentar los requisitos establecidos en dicho numeral.
9.1 Cuando el medicamento mantiene la información y características que han sido aprobadas durante la vigencia del registro, al solicitar la renovación debe presentar:
9.1.1 Comprobante de pago.
9.1.2 Solicitud de renovación de registro sanitario firmada y sellada por el Profesional Responsable, conteniendo la información detallada en el Anexo 2 de este reglamento.
9.1.3 Declaración jurada emitida por el titular o su representante legal o por el profesional responsable del registro mediante poder emitido por el titular del producto, que la información y características del producto no han variado desde la última solicitud de modificación presentada ante la Autoridad Reguladora.
En caso que la declaración jurada sea emitida en el extranjero, debe venir debidamente legalizada.
9.1.4 Certificado de Producto Farmacéutico tipo OMS o en su defecto Certificado de Libre Venta y Certificado de Buenas Prácticas de Manufactura de conformidad a lo establecido en el numeral 7.3, debiendo estar vigente en el momento de su presentación.
9.1.5 Etiquetado del producto tal y como se está comercializando, en original conforme al Reglamento Técnico Centroamericano de Etiquetado de Productos Farmacéuticos para uso Humano vigente.
NOTA: Cuando el producto no ha sido comercializado, se aceptará el proyecto del arte de los textos de impresión del empaque primario y secundario e inserto en idioma espańol, acompańado de una declaración jurada del titular del producto que indique que el producto no ha sido comercializado.
9.1.6 Informe del Estudio de Estabilidad según Reglamento Técnico Centroamericano vigente, firmado por el responsable técnico del estudio de estabilidad así designado por el titular. Este requisito rige sólo para productos que en anteriores registros sanitarios o renovaciones no hayan presentado un estudio a largo plazo.
9.2 En los casos en que el medicamento presente modificaciones en el registro sanitario y que no sean de conocimiento de la Autoridad Reguladora, podrán solicitarse en forma simultánea a la renovación. De igual manera, sino se puede presentar la declaración jurada, en ambos casos deberán cumplir los siguientes requisitos:
9.2.1 Comprobante de pago.
9.2.2 Solicitud de renovación de registro sanitario y los cambios no presentados, firmada y sellada por el Profesional Responsable.
9.2.3 Certificado de Producto Farmacéutico tipo OMS o en su defecto Certificado de Libre Venta y Certificado de Buenas Prácticas de Manufactura de conformidad a lo establecido en el numeral 7.3, debiendo estar vigente en el momento de su presentación.
9.2.4 Etiquetado del producto tal y como se está comercializando, en original conforme al Reglamento Técnico Centroamericano de Etiquetado de Productos Farmacéuticos para uso Humano vigente.
NOTA: Cuando el producto no ha sido comercializado, se aceptará el proyecto del arte de impresión del empaque primario y secundario e inserto en idioma espańol, acompańado de una declaración jurada del titular del producto que indique que el producto no ha sido comercializado.
9.2.5 Informe del Estudio de Estabilidad según Reglamento Técnico Centroamericano vigente, firmado por el responsable técnico del estudio de estabilidad así designado por el titular. Este requisito rige sólo para productos que en anteriores registros sanitarios o renovaciones no hayan presentado un estudio a largo plazo.
9.2.6 Fórmula cuantitativa y cualitativa deberá presentarse de conformidad a los requisitos establecidos en el numeral 7.5.
9.2.7 Especificaciones organolépticas, físicas, químicas, biológicas y microbiológicas del producto terminado que cumplan con lo establecido en el Reglamento Técnico Centroamericano de Verificación de la Calidad vigente. Los medicamentos con concentraciones mayores del 30% de alcohol; así como los que por su naturaleza son antisépticos, quedan exentos de presentar especificaciones microbiológicas.
9.2.8 Poderes que acrediten la representación legal y/o técnica otorgada por el titular a la(s) persona(s) natural (les) o jurídica(s), de acuerdo a la legislación de cada país; en caso de que no conste en el expediente o que haya cambios en la designación.
9.2.9 Contrato de fabricación o en su defecto el extracto relativo a las partes del contrato de fabricación, cuando aplique, según lo establecido en el numeral 7.4.
9.2.10 Según la modificación solicitada deberá presentar los documentos según Anexo 1.
10. VIGENCIA DEL REGISTRO SANITARIO
El Registro Sanitario tendrá un periodo de vigencia de 5 ańos contados a partir de su otorgamiento, pudiendo ser renovado por períodos similares. En casos de infracciones a las normas y leyes sanitarias o reglamentarias, la Autoridad Reguladora procederá a la cancelación del mismo.
11. CAUSAS DE NO OTORGAMIENTO DEL REGISTRO SANITARIO
11.1 Que exista discrepancia entre el resultado analítico y la documentación presentada. Esta causa no aplicará para el caso de Costa Rica.
11.2 Que carezca de eficacia terapéutica o seguridad según la literatura de referencia.
11.3 Que los estudios o investigaciones que se presenten en apoyo de la solicitud sean incompletos, o insuficientes para demostrar la calidad, seguridad y eficacia del producto.
11.4 Que la documentación presentada según la reglamentación vigente estén incompletos, incorrectos o no vigentes.
12. CAUSAS DE CANCELACIÓN DEL REGISTRO SANITARIO
12.1 Que el producto resulte ser nocivo o no-seguro en las condiciones normales de uso, siguiendo el debido proceso de acuerdo a la legislación de cada país.
12.2 Que se haya demostrado que el producto no es terapéuticamente eficaz.
12.3 Cuando se demuestre que el producto no tiene la composición cuantitativa o cualitativa autorizada o cuando se incumplan las garantías de calidad y estabilidad, declaradas en el expediente, siguiendo el debido proceso de acuerdo a la legislación de cada país.
12.4 Que se demuestre que los datos e información contenidos en el expediente de registro, son erróneos o falsos.
12.5 Que previo apercibimiento, se siga incumpliendo el Reglamento Técnico Centroamericano de Etiquetado de Medicamentos vigente.
12.6 Que por cualquier otra causa justificada constituya un riesgo previsible para la salud o seguridad de las personas.
12.7 Cuando se compruebe falsedad en la declaración jurada presentada para renovación del registro.
12.8 Cuando el titular del registro lo solicite.
13. EXCEPCIONES AL REGISTRO SANITARIO
La Autoridad Reguladora podrá autorizar la importación y utilización de medicamentos sin registro sanitario en los siguientes casos:
13.1 Donaciones
13.2 Emergencias nacionales y necesidad pública declaradas oficialmente.
13.3 Medicamentos huérfano& para los Estados Partes.
13.4 Medicamentos utilizados en estudios clínicos con protocolos aprobados
13.5 En casos de justificación médica.
13.6 Muestras para realizar trámites de registro.
13. 7 Medicamentos comprados a través del Fondo Rotatorio OPS.
14. MODIFICACIONES POSTERIORES AL REGISTRO SANITARIO
Toda modificación en la información que se haga posterior al registro sanitario, deberá ajustarse a lo establecido en la Clasificación y Requisitos (Anexo 1).
15. DEROGACIONES
Este Reglamento Técnico Centroamericano deroga solamente lo dispuesto en los requisitos para registro Sanitario, renovación y modificaciones de medicamentos para uso humano de la reglamentación interna de cada estado parte, a excepción de lo establecido en propiedad intelectual y bioquivalencia.
16. VIGILANCIA Y VERIFICACIÓN
Corresponde la vigilancia y verificación de este Reglamento Técnico Centroamericano a las Autoridades Reguladoras de los Estados Parte de acuerdo a su legislación.
ANEXO 1
(Normativo)
CLASIFICACIÓN Y REQUISITOS PARA LAS MODIFICACIONES AL REGISTRO SANITARIO
El Profesional Responsable podrá solicitar cualquier cambio postregistro si el poder otorgado por el titular del producto le otorga esa potestad.
A. Modificaciones que requieren aprobación previa por la Autoridad Reguladora.





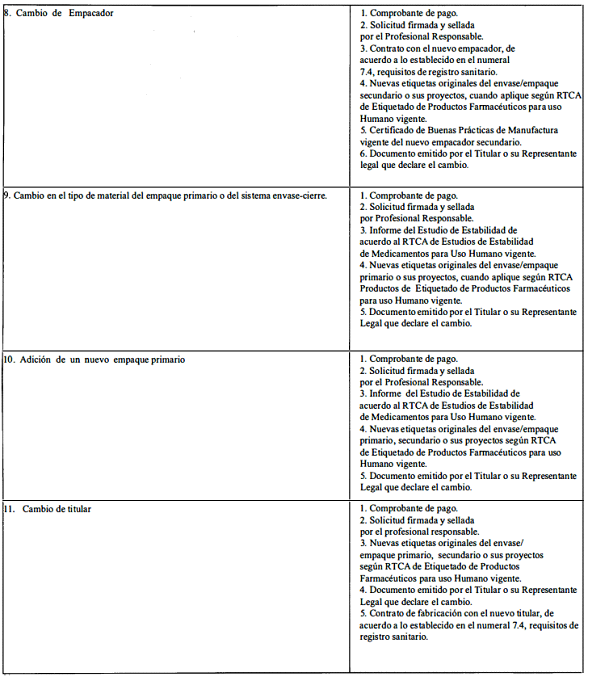
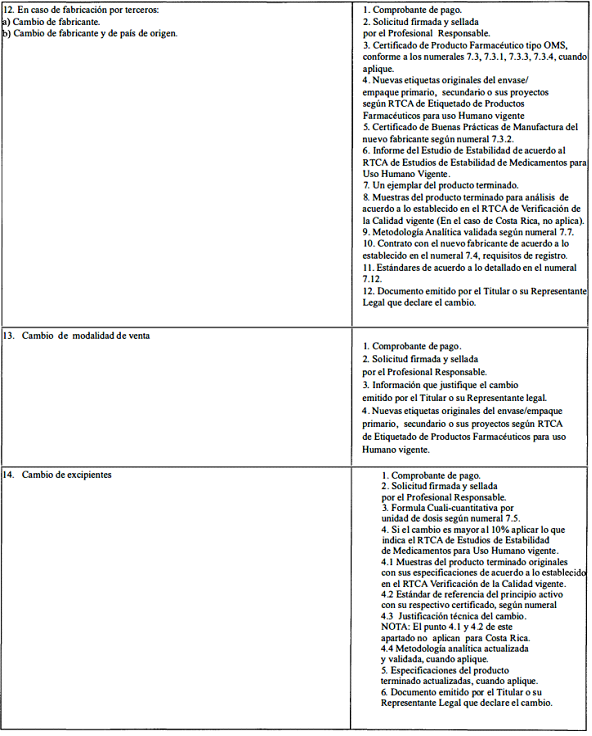
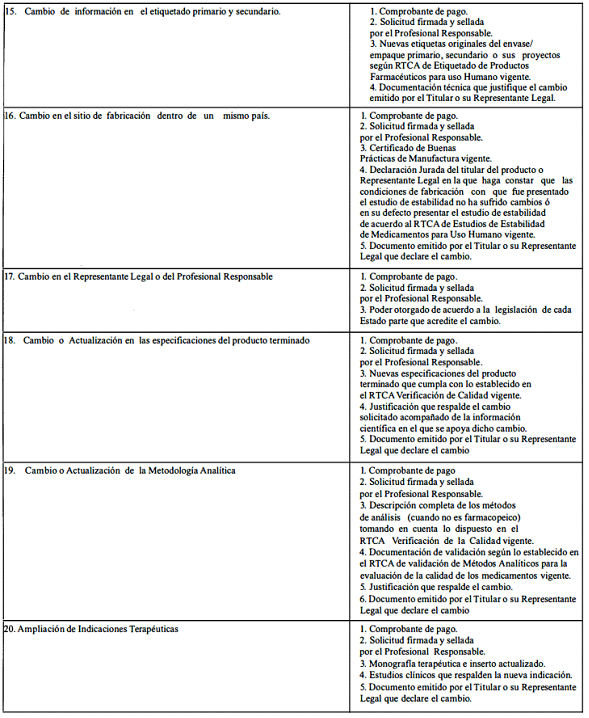
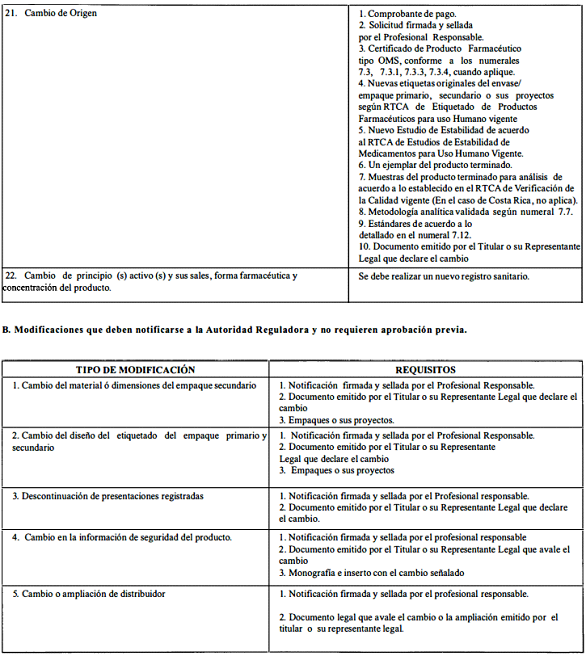
ANEXO 2
(NORMATIVO)
INFORMACIÓN A INCLUIR EN LA SOLICITUD DE REGISTRO SANITARIO.
1. Datos del producto
1.1 Nombre del producto
1.2 Nombre de los principios activos cuando contenga uno o dos principios activos
1.3 Forma farmacéutica
1.4 Vía de administración
1.5 Presentación del producto
1.6 Vida útil propuesta y condiciones de almacenamiento.
1.7 Grupo terapéutico.
1.8 Modalidad de venta.
1.9 Tipo de producto (innovador, multiorigen, etc.).
1.10 Categoría de registro (nuevo, renovación).
1.11 Metodología analítica (farmacopeica y no farmacopeica).
1.12 Estándar de referencia, cuando aplique.
2. Datos del fabricante y acondicionador:
2.1 Nombre del o de los laboratorios que participan en la fabricación.
2.2 Dirección, teléfono, fax y correo electrónico.
2.3 Etapa de fabricación.
2.4 País de laboratorio fabricante.
2.5 Número de Licencia Sanitaria o Permiso Sanitario de Funcionamiento y fecha de vencimiento (cuando sea nacional).
3. Datos del Titular del Producto:
3.1 Nombre.
3.2 Dirección, teléfono, fax y correo electrónico.
3.3 País.
4. Datos del o los Distribuidores:
4.1 Nombre del o de los distribuidores.
4.2 Dirección, teléfono, fax y correo electrónico.
4.3 Número de Licencia Sanitaria o Permiso Sanitario de Funcionamiento y fecha de vencimiento.
Nota: Para Honduras, El Salvador, estos datos son optativos.
5. Datos del Representante Legal:
5.1 Nombre.
5.2 Número de documento de identidad.
5.3 Dirección, teléfono, fax y correo electrónico.
6. Datos de profesionales Responsable:
6.1 Nombre.
6.2 Número de documento de identidad.
6.3 Dirección, teléfono, fax y correo electrónico.
6.4 Número Colegiado ó de Inscripción Químico Farmacéutico.
7. Leyenda que le dé carácter de Declaratoria Jurada a la solicitud.ANEXO 2 DE LA RESOLUCIÓN No. 333-2013
(COMIECO-LXVI)
RECONOCIMIENTO MUTUO DE REGISTRO SANITARIO DE MEDICAMENTO PARA USO HUMANO
ÁMBITO DE APLICACIÓN
El Reconocimiento aplica a todo registro sanitario de medicamentos originarios de los Estados Partes
1. REQUISITOS
a. Comprobante de pago
b. Solicitud para el reconocimiento de registro firmada y sellada por el Profesional Responsable y el Titular ó su Representante Legal, ante las Autoridades Reguladoras de los Estados Partes.
c. Poder debidamente legalizado que acredite la representación legal o técnica otorgada por el titular a la persona física o jurídica que resida en forma permanente en el país donde se solicite el reconocimiento mutuo. En caso que el Representante Legal posea la facultad podrá otorgar el poder al Profesional Responsable.
d. Certificado de Producto Farmacéutico original, emitido por el país de origen, debidamente legalizado, incluya la formula cuali-cuantitativa, el tiempo de vida útil, las condiciones de almacenamiento aprobadas, la modalidad de venta aprobada del producto y el cumplimiento de las Buenas Prácticas de Manufactura del laboratorio fabricante. Cuando en el proceso de fabricación se involucren dos o más laboratorios, la identificación de los mismos y el cumplimiento de las Buenas Prácticas de Manufactura deben incluirse como anexo.
e. El interesado presentará una copia del expediente completo junto con una declaración jurada donde indique que es copia fiel del presentado en el país en donde se realizo el registro, con la finalidad de contar con la información necesaria para realizar la vigilancia sanitaria posterior al Reconocimiento. Este requisito se presentará al momento de la entrega del documento de aprobación de Reconocimiento.
2. PROCEDIMIENTO
a. Presentación de los requisitos establecidos ante la Autoridad Reguladora.
b. La Autoridad Reguladora verifica los requisitos presentados.
c. La Autoridad Reguladora resuelve en un término de 8 días hábiles, emitiendo el respectivo documento conforme al Anexo II.
d. En caso de aprobación se asignará el código RM e inicial del país que realiza el reconocimiento y que antecede al número correlativo otorgado por el Estado Parte, el cual se conservará al momento de la renovación. Dicho código deberá incluirse de la manera que se establece para el número de registro sanitario en el RTCA de Etiquetado de Medicamento vigente. La vigencia del reconocimiento será la misma del registro original.
3. RECONOCIMIENTO A LAS MODIFICACIONES DEL REGISTRO
a. Solicitud para el reconocimiento de la modificación del registro firmada y sellada por el Profesional Responsable y el Titular ó su Representante Legal, ante las Autoridades Reguladoras de los Estados Partes.
b. Copia de la documentación que respalde el cambio de conformidad con el Anexo 1 del RTCA Productos Farmacéuticos Medicamentos para uso Humano Requisitos de Registro Sanitario, con la finalidad de contar con la información necesaria para realizar la vigilancia sanitaria. Este requisito se presentará al momento de la entrega del documento de aprobación de Reconocimiento.
c. Documento de aprobación del cambio.
3.2 PROCEDIMIENTO
a. Presentación de los requisitos establecidos ante la Autoridad Reguladora.
b. La Autoridad Reguladora verifica los requisitos presentados.
c. La Autoridad Reguladora resuelve en un término de 8 días hábiles, emitiendo el respectivo documento conforme el Anexo II.
d. En caso de aprobación el interesado entrega la documentación de respaldo, en el momento de recibir dicho documento.
4. RENOVACIÓN DEL RECONOCIMIENTO AL REGISTRO
4.1 REQUISITOS
a. Comprobante de pago
b. Solicitud para la renovación del reconocimiento de registro firmada y sellada por el Profesional Responsable y el Titular ó su Representante Legal, ante la Autoridad Reguladora de los Estados Partes.
c. Certificado de Producto Farmacéutico original, emitido por el país de origen, debidamente legalizado, que incluya la formula cuali-cuantitativa, el tiempo de vida útil, las condiciones de almacenamiento aprobadas, la modalidad de venta aprobada del producto y el cumplimiento de las Buenas Prácticas de Manufactura del laboratorio fabricante. Cuando en el proceso de fabricación se involucren dos o más laboratorios, la identificación de los mismos y el cumplimiento de las Buenas Prácticas de Manufacturas deben incluirse como anexo.
4.1.1 Si al momento de realizar la renovación del reconocimiento hubiese modificaciones o cambios en el registro original que no se hayan notificados debe presentar:
1. Solicitud de renovación de reconocimiento que incluya los cambios post registro no notificados
2. Documento de aprobación del cambio.
3. Copia de la documentación que respalde el cambio de conformidad con el Anexo 1 del RTCA Producto Farmacéutico Medicamentos para uso humano Requisitos de Registro Sanitario, con la finalidad de contar con la información necesaria para realizar la vigilancia sanitaria. Este requisito se presentará al momento de la entrega del documento de aprobación de Reconocimiento.
4.2 PROCEDIMIENTO
a. Presentación de los requisitos establecidos ante la Autoridad Reguladora.
b. La Autoridad Reguladora verifica los requisitos presentados.
c. La Autoridad Reguladora resuelve en un término de 8 días hábiles, emitiendo el respectivo documento conforme al Anexo II.
d. En caso de aprobación se debe mantener el número de reconocimiento y vigencia otorgada en la renovación del registro.
e. El interesado entrega la documentación que respalda el cambio, en el momento de recibir la aprobación de la renovación.
5. CAUSA DE NO RECONOCIMIENTO
No se otorgará reconocimiento al registro cuando:
a. Exista confusión o igualdad en el nombre comercial de un producto farmacéutico previamente registrado.
b. El medicamento contenga ingredientes activos o combinaciones de los mismos, que no cuenten con evidencia científica documentada de su seguridad y eficacia.
c. En una formulación donde se combinan principios activos de síntesis química con productos naturales medicinales, y dicha combinación no esté catalogada como medicamento.
d. La formulación sea un suplemento nutricional o productos naturales medicinales.
e. Exista una alerta internacional que cuestione la seguridad y eficacia del principio activo o combinaciones de los mismos.
f. En caso de coempaque que no se justifique científicamente para el esquema de tratamiento solicitado.
g. Si la modalidad de venta aprobada en el registro del país de origen difiere a la del país de reconocimiento.
6. CAUSAS DE CANCELACIÓN
a. Que el producto resulte nocivo o no-seguro en las condiciones normales de uso.
b. Que se haya demostrado con evidencia científica concluyente que el producto no es terapéuticamente eficaz.
c. Cuando se demuestre que el producto no tiene la composición cuantitativa o cualitativa autorizada o cuando se incumpla las garantías de calidad y estabilidad, declaradas en el expediente.
d. Que se demuestre falsedad en los datos e información contenidos en el expediente presentado para Reconocimiento Mutuo.
e. Que previo apercibimiento, el etiquetado con el que se comercialice el producto en el país del reconocimiento sea diferente al etiquetado aprobado en el registro original.
f. Que por cualquier otra causa justificada constituya un riesgo previsible para la salud o seguridad de las personas.
g. Cuando la Autoridad Reguladora que otorgó el registro sanitario original notifique la cancelación del mismo.
h. Cuando el titular del reconocimiento al registro lo solicite.
7. RECONOCIMIENTO DE PRODUCTOS COEMPACADOS
En caso que se realice una solicitud de reconocimiento para medicamento coempacados deberá presentar:
a. Comprobante de pago
b. Solicitud para el reconocimiento de coempaque firmada y sellada por el Profesional Responsable y el Titular ó su Representante Legal, ante las Autoridades Reguladoras de los Estados Partes.
c. Certificado de producto farmacéutico que incluya, para cada uno de los productos del coempaque la formula cuali-cuantitativa, el tiempo de vida útil, las condiciones de almacenamiento aprobadas, la modalidad de venta aprobada del producto y el cumplimiento de las Buenas Prácticas de Manufacturas del laboratorio fabricante. Cuando en el proceso de fabricación se involucren dos o más laboratorios, la identificación de los mismos y el cumplimiento de las Buenas Prácticas de Manufacturas deben incluirse como anexo.
d. Documento de aprobación del coempaque emitido por la Autoridad Reguladora del país del registro original.
e. El interesado presentará una copia del expediente completo junto con una declaración jurada donde indique que el documento presentado es copia fiel del presentado en el país en donde se realizó el registro, con la finalidad de contar con la información necesaria para realizar la vigilancia sanitaria posterior al reconocimiento. Este requisito se presentará al momento de la entrega del documento de aprobación de reconocimiento.
f. Copia del proyecto o empaque original, aprobados.
8. DISPOSICIONES GENERALES
a. Para Costa Rica el Procedimiento de Reconocimiento Mutuo aplica para los medicamentos que hayan sido registrados con la reglamentación armonizada vigente.
b. Para los productos del CA4 que a la fecha de entrada en vigencia del presente procedimiento hayan sido reconocidos como Medicamentos y estén clasificados como suplemento nutricional o producto natural no serán renovados.
c. No se podrá comercializar un producto sin haber notificado las modificaciones realizadas al registro original.
d. Para los productos sujetos a protección de datos de prueba, se aplicará la normativa vigente de cada país.
e. Los productos que requieran estudios de bioequivalencia quedan sujetos a la normativa vigente de cada país.
f. Este procedimiento no aplica para los productos biológicos y biotecnológicos.
ANEXO I
(INFORMATIVO)
INFORMACIÓN A INCLUIR
EN LA SOLICITUD DE RECONOCIMIENTO
1. Tipo de trámite
1.1 Reconocimiento
1.2 Renovación
1.3 Modificación
1.4 Coempaque
2. Datos del producto
2.1 Nombre del producto
2.2 Nombre del o los principios activos con su denominación común internacional.
2.3 Forma farmacéutica.
2.4 Vía de administración
2.5 Presentación del producto
2.6 Vida útil aprobada y condiciones de almacenamiento
2.8 Grupo terapéutico
2.7 Modalidad de venta.
3. Datos del fabricante y acondicionador:
3.1 Nombre del ó de los laboratorios que participan en la fabricación y acondicionamiento del producto.
3.2 Dirección, teléfono, fax y correo electrónico.
3.3 Etapa de proceso de fabricación.
3.4 País del o de los laboratorios que participan en la fabricación y acondicionamiento del producto.
4. Datos del titular del producto:
4.1 Nombre
4.2 Dirección, teléfono, fax y correo electrónico.
4.3 País.
5. Datos del o los distribuidores
5.1 Nombre del o de los distribuidores
5.2 Dirección, teléfono, fax y correo electrónico.
5.3 Número de licencia sanitaria y fecha de vencimiento.
6. Datos del representante legal:
6.1 Nombre
6.2 Número de documento de identidad.
6.3 Dirección, teléfono, fax y correo electrónico.
7. Datos de persona física/natural o jurídica registrante
7.1 Nombre
7.2 Número de documento de identidad.
7.3 Dirección, teléfono, fax y correo electrónico.
8. Datos de profesional responsable
8.1 Nombre
8.2 Número de documento de identidad
8.3 Dirección, teléfono, fax y correo electrónico.
8.4 Número colegiado ó de inscripción químico farmacéutico.
ANEXO II
INFORMACIÓN A INCLUIR EN EL DOCUMENTO DE APROBACIÓN EMITIDO POR LA AUTORIDAD
REGULADORA DE LOS ESTADOS PARTES A LA SOLICITUD DE RECONOCIMIENTO
IDENTIFICACIÓN DE LA AUTORIDAD REGULADORA
QUE APRUEBA O RECHAZA EL RECONOCIMIENTO
Con fundamento en lo dispuesto en la Resolución COMIECO No. 333-13 se reconoce el (Registro Sanitario, Renovación de Registro, Modificaciones al Registro Sanitario, Co. Empaque) al MEDICAMENTO otorgado por la Autoridad Reguladora de:______________________________________
Al producto: ___________________________________________________________________________
Nombre genérico: ______________________________________________________________________
Vía de administración: ___________________________________________________________________
Forma farmacéutica: ____________________________________________________________________
Concentración por unidad posológica:_______________________________________________________
Presentación del producto:________________________________________________________________
_____________________________________________________________________________________
Vida útil aprobada:______________________________________________________________________
Condiciones de almacenamiento:__________________________________________________________
Nombre del titular del registro:___________________________________________País______________
Nombre del fabricante:___________________________________________________País____________
Modalidad de venta:_____________________________________________________________________
Número de reconocimiento de registro sanitario:_______________________________________________
Vigencia:______________________________________________________________________________
Firma de la Autoridad Reguladora y sello:________________________________________________
-FIN DEL DOCUMENTO-