Enlace a Legislación Relacionada
(REGLAMENTO TÉCNICO CENTROAMERICANO RTCA 65.05.51:18 MEDICAMENTOS VETERINARIOS, PRODUCTOS AFINES Y SUS ESTABLECIMIENTOS. REQUISITOS DE REGISTRO SANITARIO Y CONTROL)
RESOLUCIÓN N°. 436-2020 (COMIECO-XCIII), aprobada el 10 de diciembre de 2020
Publicada en La Gaceta, Diario oficial N°. 122 del 02 de julio de 2021
EL CONSEJO DE MINISTROS DE INTEGRACIÓN ECONÓMICA
CONSIDERANDO:
Que de conformidad con los artículos 38, 39 y 55 del Protocolo al Tratado General de Integración Económica Centroamericana (Protocolo de Guatemala), modificado por la Enmienda del 27 de febrero de 2002, el Consejo de Ministros de Integración Económica (COMIECO), tiene bajo su competencia los asuntos de la Integración Económica Centroamericana y, como tal, le corresponde aprobar los actos administrativos aplicables en los Estados Parte del Subsistema Económico; Que según los artículos 7 y 26 del Protocolo de Guatemala, los Estados Parte han convenido establecer un proceso de armonización regional de la normativa técnica;
Que el COMIECO, mediante la Resolución No. 257-2010 (COMIECO-LIX), del 13 de diciembre de 2010, aprobó el Reglamento Técnico Centroamericano RTCA 65.05.51:08 Medicamentos Veterinarios y Productos Afines. Requisitos de Registro Sanitario y Control, el cual se modificó parcialmente mediante los actos administrativos siguientes: Resolución No. 326-2013 (COMIECO-LXVI), del 12 de diciembre de 2013; Resolución No. 360-2014 (COMIECO-LXX), del 4 de diciembre de 2014; y la Resolución No. 362-2015 (COMIECO-LXXI), del 23 de abril de 2015;
Que las instancias de la Integración Económica tomaron la decisión de revisar el RTCA 65.05.51:08 Medicamentos Veterinarios y Productos Afines. Requisitos de Registro Sanitario y Control y sus modificaciones, para ajustarlo en virtud de la experiencia adquirida y las mejores prácticas internacionales aplicables;
Que los Estados Parte, en su calidad de Miembros de la Organización Mundial del Comercio (OMC), notificaron al Comité de Obstáculos Técnicos al Comercio y al Comité de Medidas Sanitarias y Fitosanitarias, de conformidad con lo establecido en el Acuerdo sobre la Aplicación de Medidas Sanitarias y Fitosanitarias y el Acuerdo sobre Obstáculos Técnicos al Comercio, el Proyecto de Reglamento Técnico Centroamericano RTCA 65.05.51:18 Medicamentos Veterinarios, Productos Afines y sus Establecimientos. Requisitos de Registro Sanitario y Control;
Que los Estados Parte, concedieron un plazo prudencial a los Estados Miembros de la OMC para hacer observaciones al Proyecto referido, según lo establecido en el numeral 4), párrafo 9 del artículo 2 del Acuerdo sobre Obstáculos Técnicos al Comercio, observaciones que fueron debidamente analizadas y atendidas en lo pertinente;
Que de conformidad con el artículo 2, párrafo 12 del Acuerdo sobre Obstáculos Técnicos al Comercio de la OMC, los Miembros preverán un plazo prudencial entre la aprobación de los reglamentos técnicos y su entrada en vigor, con el fin de dar tiempo a los productores para adaptar sus productos o sus métodos de producción a lo establecido en los reglamentos técnicos;
Que en cumplimiento con el artículo 55, párrafo 3, del Protocolo de Guatemala, el Reglamento se remitió a consulta del Comité Consultivo de la Integración Económica (CCIE);
Que el COMIECO se puede reunir de manera virtual mediante el sistema de vídeo conferencia, en cuyo caso, le corresponde a la Secretaría de Integración Económica Centroamericana (SIECA) recopilar la firma de cada uno de los Ministros o Viceministros, según corresponda, en su respectivo país,
POR TANTO:
Con fundamento en los artículos 1, 3, 5, 7, 15, 26, 36, 37, 38, 39, 46, 52 y 55 del Protocolo de Guatemala; y 19, 20 Bis, 32 y 32 Bis del Reglamento de Organización y Funcionamiento de los Consejos: de Ministros de Integración Económica, Intersectorial de Ministros de Integración Económica y Sectorial de Ministros de Integración Económica,
RESUELVE:
1. Modificar, por sustitución total, el Reglamento Técnico Centroamericano RTCA 65.05.51 :08 Medicamentos Veterinarios y Productos Afines. Requisitos de Registro Sanitario y Control, aprobado mediante la Resolución No. 257-2010 (COMIECO-LIX), del 13 de diciembre de 2010; por el Reglamento Técnico Centroamericano RTCA 65.05.51:18 Medicamentos Veterinarios, Productos Afines y sus Establecimientos. Requisitos de Registro Sanitario y Control, en la forma que aparece en el Anexo de la presente Resolución y que forma parte integrante de la misma.
2. Los Estados Parte deberán implementar el Código de Buenas Prácticas de Manufactura (BPM) de Medicamentos Veterinarios del Comité para las Américas de Medicamentos Veterinarios de la Organización Mundial de Sanidad Animal, así como su Guía de Verificación, ambos en su versión vigente, en un plazo de cinco (5) años, a partir de la fecha de entrada en vigor del RTCA 65.05.51:18.
Durante el plazo de 5 años indicado en el párrafo anterior, cuando un fabricante de un Estado Parte solicite el registro sanitario o la renovación de su registro en otro Estado Parte, presentará el documento oficial, expedido por su Autoridad Competente, que respalde la implementación del plan operativo para el cumplimiento de las BPM.
3. En el caso de Guatemala, El Salvador, Honduras, Nicaragua y Costa Rica, el titular del registro sanitario deberá cumplir con la codificación establecida en el Anexo H del RTCA 65 .05.51:18, en un plazo de veinticuatro (24) meses, contados a partir de la entrada en vigor del RTCA 65.05.51: 18. En el caso de Panamá, dicho plazo será de treinta (30) meses, a partir de la entrada en vigor del RTCA 65.05.51:18. Durante estos plazos, el titular del registro sanitario podrá comercializar los productos bajo la codificación vigente. Una vez vencidos dichos plazos, el titular del registro sanitario deberá comercializar todos sus productos con la codificación que establece el Anexo H del RTCA 65 .05.51:18.
4. Derogar los actos administrativos siguientes:
a. Resolución No. 257-2010 (COMIECO-LIX), del 13 de diciembre de 2010;
b. Resolución No. 326-2013 (COMIECO-LXVI), del 12 de diciembre de 2013;
c. Resolución No. 360-2014 (COMIECO-LXX), del 4 de diciembre de 2014; y,
d. Resolución No. 362-2015 (COMIECO-LXXI), del 23 de abril de 2015.
5. La presente Resolución entrará en vigor el 10 de agosto de 2021 y será publicada por los Estados Parte.
Centroamérica, 10 de diciembre de 2020
(f) Duayner Salas Chaverri, Viceministro, en representación del Ministro de Comercio Exterior de Costa Rica. (f) Miguel Ángel Corleto Urey, Viceministro, en representación de la Ministra de Economía de El Salvador. (f) Edith Flores de Molina, Viceministra, en representación del Ministro de Economía de Guatemala. (f) David Antonio Alvarado Hernández, Subsecretario, en representación de la Designada Presidencial y Encargada de la Secretaría de Estado en el Despacho de Desarrollo Económico de Honduras. (f) Orlando Solórzano Delgadillo, Ministro de Fomento, Industria y Comercio de Nicaragua. (f) Juan Carlos Sosa, Viceministro, en representación del Ministro de Comercio e Industrias de Panamá.
ANEXO DE LA RESOLUCIÓN No. 436-2020 (COMIECO-XCIII)
REGLAMENTO TÉCNICO CENTROAMERICANO RTCA 65.05.51:18 ICS 65.020.30
1ra Revisión
MEDICAMENTOS VETERINARIOS, PRODUCTOS AFINES Y SUS ESTABLECIMIENTOS. REQUISITOS DE REGISTRO SANITARIO Y CONTROL
CORRESPONDENCIA: Este Reglamento no tiene correspondencia con ninguna norma internacional.
Reglamento Técnico Centroamericano, editado por:
-Ministerio de Economía, MINECO
-Organismo Salvadoreño de Reglamentación Técnica, OSARTEC
- Secretaría de Desarrollo Económico, SDE
- Ministerio de Fomento, Industria y Comercio, MIFIC
- Ministerio de Economía Industria y Comercio, MEIC
- Ministerio de Comercio e Industrias, MICI
INFORME
Los respectivos Comités Técnicos de Reglamentación Técnica a través de los entes de Reglamentación Técnica de los países centroamericanos, son los organismos encargados de realizar el estudio o la adopción de los Reglamentos Técnicos Centroamericanos. Están conformados por representantes de los sectores Académico, Consumidor, Empresa Privada y Gobierno.
Este Reglamento Técnico Centroamericano RTCA Medicamentos Veterinarios, Productos Afines y sus Establecimientos. Requisitos de Registro Sanitario y Control, en su versión vigente, fue acordado por el Subgrupo de Insumos Agropecuarios y el Subgrupo de Medidas de Normalización. La oficialización de este reglamento técnico conlleva la aprobación por el Consejo de Ministros de Integración Económica Centroamericana (COMIECO).
MIEMBROS PARTICIPANTES DEL COMITÉ
Por Guatemala
Ministerio de Agricultura, Ganadería y Alimentación (MAGA-VISAR)
Por El Salvador
Ministerio de Agricultura y Ganadería (MAG)
Por Nicaragua
Instituto de Protección y Sanidad Agropecuaria (IPSA)
Por Honduras
Secretaria de Agricultura y Ganadería (SEN-ASA)
Por Costa Rica
Ministerio de Agricultura y Ganadería (MAG-SENASA)
Por Panamá
Ministerio de Desarrollo Agropecuario (MIDA)1. OBJETO
Establecer las disposiciones de registro sanitario y control de los medicamentos para uso veterinario, productos afines y sus establecimientos.2. ÁMBITO DE APLICACIÓN
Aplica a los medicamentos veterinarios y productos afines, así como a los establecimientos que los registran, fabrican, comercializan, despachan o expenden, fraccionan o almacenan en los Estados Parte.
Se exceptúa del ámbito de aplicación de este reglamento el registro sanitario de las materias primas para elaborar medicamentos veterinarios y productos afines, así como las formulaciones magistrales veterinarias y sus establecimientos, lo cual queda sujeto a la legislación nacional de cada Estado Parte.
NOTA 1. Las disposiciones del presente reglamento deben interpretarse en beneficio y protección de la salud humana, la salud animal, el medio ambiente y el patrimonio pecuario en cada uno de los Estados Parte, para cumplir con los objetivos institucionales que cada uno de los Estados Parte tenga en sus distintos cuerpos normativos.
3. DEFINICIONES
Para los efectos de este reglamento, se establecen las siguientes definiciones:
3.1 Almacenadores: establecimientos dedicados a almacenar medicamentos veterinarios y productos afines, de fabricantes o comercializadores debidamente registrados y aprobados por la autoridad competente.
3.2 Anotación marginal o modificación de registro sanitario: cambio de un registro sanitario original, avalado por la autoridad competente a solicitud del titular.
3.3 Autoridad competente: entidad encargada de la aplicación del presente reglamento, para su efectivo cumplimiento por los sectores involucrados en el tema y actividad que éste comprende.
3.4 Certificado de análisis: documento emitido por el laboratorio de control de calidad del fabricante u otro oficialmente autorizado, que certifica los resultados obtenidos del análisis de calidad de un lote específico.
3.5 Certificado de libre venta: documento oficial, emitido por la autoridad competente del país de origen del producto, en el cual se certifica que un medicamento veterinario o producto afín es comercializado en su territorio según las condiciones de venta establecidas.
3.6 Certificado de registro sanitario: documento oficial emitido por la autoridad competente que da fe que ha cumplido con todos los requisitos establecidos para el registro sanitario.
3.7 Envase o empaque: todo recipiente o envoltura destinada a conservar la calidad e inocuidad del medicamento veterinario o producto afín, facilitando su manipulación.
3.8 Envase o empaque primario: recipiente dentro del cual se coloca directamente el medicamento veterinario o producto afín en su forma farmacéutica terminada con el propósito de protegerlo de su deterioro, contaminación o adulteración y facilitar su manejo.
3.9 Envase o empaque secundario: recipiente dentro del cual se coloca el envase primario que contiene al medicamento veterinario o producto afín en su forma farmacéutica terminada, para su distribución y comercialización.
3.10 Estado Parte: Estados que son parte del Protocolo al Tratado General de Integración Económica Centroamericana - Protocolo de Guatemala.
3.11 Estándar analítico: sustancia de pureza y calidad conocidas, utilizada como patrón de comparación (primario o secundario) en los ensayos de laboratorio de control de calidad.
3.12 Estándar primario: aquella sustancia que ha demostrado a través de una serie de análisis, ser un material de alta pureza obtenido de una fuente oficial reconocida y cuyo contenido asignado es aceptado sin requerir comparación con otra sustancia química.
NOTA 1. Las sustancias farmacopeicas de referencia química son consideradas como estándar primario.
3.13 Estándar secundario: sustancia de calidad y pureza establecida, la cual es comparada con un estándar de referencia primario, usado para análisis rutinario del laboratorio.
3.14 Establecimiento veterinario: espacio físico donde se fabrican, comercializan, despachan o expenden, fraccionan, gestionan registros o almacenan medicamentos veterinarios y productos afines.
3.15 Etiquetado: toda información que se adhiera, imprima o grabe en el envase y empaque de presentación comercial de un medicamento veterinario o producto afín.
3.16 Excipiente, ingrediente inerte, vehículo: materia que se agrega a los principios activos o a sus asociaciones para servirles de vehículo, posibilitar su preparación, estabilidad, modificar sus propiedades organolépticas o determinar las propiedades físico-químicas del medicamento y su biodisponibilidad.
3.17 Fabricante: toda persona física (natural, individual) o jurídica legalmente constituida que se dedica a la elaboración o formulación de medicamentos veterinarios y productos afines, pudiendo ser maquiladores.
3.18 Farmacia veterinaria o expendio: establecimiento legalmente constituido que se dedica a la comercialización de medicamentos veterinarios y productos afines directamente al público.
3.19 Forma cosmética: denominación que recibe un grupo de productos que tienen características físicas comunes, por ejemplo: champú, jabón, loción y otros.
3.19 Fórmula cuali-cuantitativa: descripción completa de la composición y su contenido, incluyendo ingredientes activos e inertes, con elementos simples o compuestos, de un medicamento veterinario o producto afín, emitida por el fabricante.
3.20 Fraccionador: toda persona física (natural, individual) o jurídica legalmente constituida que se dedica a reempacar o reenvasar un medicamento veterinario o producto afín siguiendo con las buenas prácticas de manufactura.
3.21 Información falsa: aquella información que se presenta con el objetivo de sustentar un registro sanitario, que no corresponde a la verdadera información del producto y que, con intención, se hace pasar por auténtica.
3.22 Información inexacta: aquella que se presenta con el objetivo de sustentar un registro sanitario y que sin intención no es precisa.
3.23 Inserto o prospecto: instructivo impreso que acompaña a cada presentación comercial de un medicamento veterinario o producto afín, cumpliendo con las disposiciones sobre etiquetado del presente reglamento.
3.24 Literatura científica reconocida o documento técnico de respaldo: se considera como una fuente válida aquella que proviene de revistas indexadas o revisadas por pares1, fuentes o guías oficiales, farmacopeas, libros o monografías farmacopeicas cuyas citas permitan tener la trazabilidad de la fuente primaria, sin detrimento de la necesidad de aportar dicha fuente primaria cuando se requiera; estudios realizados bajo guías internacionalmente reconocidas, así como estudios privados aportados por el registrante efectuados bajo normas internacionales reconocidas.
3.25 Maquilador: toda persona física (natural, individual) o jurídica legalmente constituida que presta servicios a terceros de formulación o elaboración de un producto.
3.26 Materia prima: sustancia, cualquiera que sea su origen, activa o inactiva, utilizada como componente principal, ingrediente activo o excipiente que se usa para la fabricación de medicamentos veterinarios y productos afines, tanto si permanece inalterada como si sufre modificación.
____________
1 Revisada por pares se refiere a paneles de expertos reconocidos en la materia, los cuales verifican que la información y datos son correctos y reales, apegados a la ciencia
3.27 Medicamento innovador: es aquel medicamento que resulta de un proceso de investigación, que está protegido por una patente y es fabricado de manera exclusiva por el laboratorio farmacéutico que la desarrolló. Se denominan por el nombre de la sustancia activa y por un nombre o marca comercial.
Es el producto que fue primero autorizado para la comercialización con base en documentación de calidad, seguridad y eficacia y que ha pasado por todas las fases de desarrollo de un nuevo producto.
3.28 Medicamento genérico: medicamento veterinario que contiene el/los mismo/s principio/s activo/s, la misma sal o éster del principio activo que un producto registrado con anterioridad y que no cuenta con protección de patente; en la misma concentración, forma farmacéutica, vía de administración, posología e indicaciones terapéuticas, destinado a la misma especie y categoría, debiendo ser bioequivalente con el producto registrado con anterioridad y diferir apenas en características relativas al tamaño, presentación, periodo de validez, embalaje, etiquetado, excipientes o vehículos. En consecuencia, deben demostrar similares biodisponibilidades, aunque pueden diferir en magnitudes y perfiles temporales de sus actividades farmacológicas.
3.29 Medicamento similar: medicamento veterinario no genérico, que contiene el/los mismo/s principio/s activo/s de un medicamento registrado con anterioridad en cualquier país, a la misma concentración y en la misma forma farmacéutica, pudiendo o no variar sus excipientes, pero respetando las especificaciones y los patrones de calidad de farmacopeas reconocidas.
3.30 Medicamento veterinario: toda sustancia o sus mezclas que puedan ser aplicadas o administradas a los animales, con fines terapéuticos, profilácticos, inmunológicos, diagnósticos, eutanásicos o para modificar las funciones fisiológicas y de comportamiento. Se incluyen los ectoparasiticidas.
3.31 Medicamento veterinario en combinaciones a dosis fijas: mezcla de dos o más principios activos que están en un mismo preparado, los cuales utilizados en combinación resultan más beneficiosos que individualmente.
3.32 Medicamento veterinario y producto afín alternativo: conjunto de substancias o sus mezclas que no son parte de la medicina convencional.
3.33 Oficina registrante: establecimiento que tiene como única actividad gestionar registros de productos y establecimientos veterinarios, su renovación o modificación ante la autoridad competente.
3.34 Período de retiro o tiempo de retiro: es el período que transcurre entre la última administración o aplicación de un medicamento y la recolección de tejidos comestibles o productos provenientes de un animal tratado, que asegura que el contenido de residuos en los alimentos se ajusta al límite máximo de residuos para los medicamentos veterinarios (LMR).
3.35 Preparaciones o formulaciones magistrales: producto medicinal elaborado en una farmacia por el farmacéutico o médico veterinario especializado, para atender una prescripción o receta del médico veterinario de un paciente individual o grupo específico de animales.
3.36 Principio activo: sustancia que posee uno o más efectos farmacológicos o que sin tener dichos efectos, al ser administrada o aplicada al organismo animal, adquiere estos efectos luego de sufrir cambios en su estructura química.
3.37 Producto: medicamento veterinario o producto afín, según corresponda.
3.38 Producto afín: toda substancia, material de cualquier origen y sus mezclas que se utilicen en los animales o en su medio de vida, con fines de diagnóstico, sanidad, higiene y
cosmético.
3.39 Producto biológico: producto veterinario elaborado a partir de bacterias, virus, sueros hiperinmunes, toxinas y productos análogos de origen natural, sintético o biotecnológicos que incluyen, reactivos-diagnósticos, antitoxinas, vacunas, microorganismos vivos, microorganismos muertos y los componentes antagónicos o inmunizantes de organismos usados en el diagnóstico, tratamiento o prevención de enfermedades en los animales.
3.40 Receta controlada (retenida): receta emitida por un médico veterinario aprobado por la autoridad competente u organismo estatutario, según la legislación nacional de cada Estado Parte, para prescribir medicamentos de uso veterinario controlado.
3.41 Registrante: persona física (natural, individual) o jurídica legalmente autorizada por el propietario o titular de registro sanitario de un medicamento veterinario o producto afín para registrarlo ante la autoridad competente. El registrante puede ser el mismo titular del registro sanitario.
3.42 Registro sanitario: procedimiento mediante el cual la autoridad competente de un Estado Parte aprueba la comercialización de un medicamento veterinario, producto afín o el funcionamiento de un establecimiento veterinario, luego de cumplir con los requisitos establecidos por ésta.
3.43 Representante legal: persona física (natural, individual) o jurídica que representa al titular o propietario del registro sanitario de un producto o establecimiento veterinario y que responde ante la autoridad competente, según sea el caso, en lo que respecta a su registro sanitario.
3.44 Regente veterinario: profesional médico veterinario que, de conformidad con las disposiciones legales de cada Estado Parte, es autorizado para que cumpla con las responsabilidades de la dirección técnica, científica y profesional de los distintos establecimientos veterinarios.
3.45 Titular o propietario del registro sanitario: persona física (natural, individual) o jurídica que tiene a su favor el registro sanitario de un producto, para su comercialización.
3.46 Venta libre: medicamento que se puede comercializar sin receta médica.
4. REGISTRO SANITARIO DE ESTABLECIMIENTOS Y SU RENOVACIÓN
Todo establecimiento veterinario debe estar registrado y autorizado por la autoridad competente. Los establecimientos se clasifican en:
a) Fabricantes
b) Fraccionadores
c) Comercializadores (importadores, exportadores, droguerías o distribuidoras)
d) Farmacias o expendios veterinarios
e) Almacenadores
f) Oficinas registrantes
g) Titulares del registro no fabricante
h) Maquiladores
4.1 Requisitos Generales
a) Poseer la autorización de funcionamiento correspondiente, bajo la normativa oficial vigente en cada Estado Parte.
b) Contar con un representante legal en el país del registro sanitario.
c) Poder de representación constituido legalmente, otorgado por el titular del registro según lo establecido por la autoridad competente de cada Estado Parte.
NOTA 1. Para el caso de Guatemala, Honduras y Panamá, este requisito no será solicitado para el registro de establecimientos, pero si para el registro de productos.
d) Contar con un regente veterinario.
e) Documentos legales que respalden la constitución de la empresa en caso de la persona jurídica y documentos de identidad del solicitante en el caso de la persona física (natural, individual).
4.2 Los fabricantes. Además de los requisitos generales deben:
a) Presentar el certificado de Buenas Prácticas de Manufactura.
b) Contar con los servicios de un laboratorio de control de calidad del fabricante, u otro autorizado por la autoridad competente debiendo aportar la copia certificada de dicha autorización con los trámites consulares respectivos.
4.3 Los fraccionadores y maquiladores. Además de los requisitos generales, deben presentar el certificado de Buenas Prácticas de Manufactura.
4.4 Vigencia. El registro sanitario de establecimiento tendrá una vigencia de 5 años y para su renovación debe cumplir con los requisitos de registro sanitario establecidos en este apartado, según corresponda. Toda solicitud de renovación puede realizarse desde tres meses antes hasta su fecha de vencimiento.
4.5 Cancelación del registro sanitario. Los registros sanitarios de establecimientos pueden ser cancelados previo a su vencimiento, al comprobarse el incumplimiento de:
a) Cualquiera de las condiciones establecidas para su otorgamiento.
b) La violación de las normativas específicas en la materia de cada Estado Parte.
5. REGISTRO SANITARIO, RENOVACIÓN Y OTROS CONTROLES PARA MEDICAMENTOS VETERINARIOS Y PRODUCTOS AFINES
Todo medicamento veterinario y producto afín que se fabrique, importe, exporte, fraccione, almacene, comercialice, despache o expenda, debe estar registrado y autorizado por la autoridad competente.
5.1 Productos objeto de registro sanitario y control
La autoridad competente de cada Estado Parte debe registrar y controlar los siguientes productos:
a) Productos farmacéuticos o de medicina alternativa de uso veterinario.
b) Productos biológicos de uso veterinario.
c) Productos afines de uso veterinario.
5.2 Clasificación de los medicamentos veterinarios y productos afines
Para efectos de registro sanitario y control de los productos, estos se clasifican de acuerdo con su nivel de riesgo en cuatro grupos:
I. Medicamentos veterinarios de uso restringido indicados en la lista emitida por la Junta Internacional de Fiscalización de Estupefacientes o por los Estados Parte, de venta y despacho o expendio exclusivo en establecimientos veterinarios aprobados para tal fin por la autoridad competente mediante receta controlada (retenida).
II. Productos de uso restringido, de venta y despacho o expendio exclusivo mediante receta controlada (retenida) en establecimientos veterinarios aprobados para tal fin por la autoridad competente.
III. Productos veterinarios de venta exclusiva en establecimientos veterinarios que cuentan con un regente médico veterinario y aprobados por la autoridad competente.
IV. Medicamentos veterinarios y productos afines de libre venta en cualquier establecimiento autorizado.
5.3 Tipos de registro sanitario
Se consideran dos tipos de registro sanitario de medicamentos veterinarios y productos afines
a) Registro sanitario común.
b) Registro sanitario simplificado.
5.3.1 Registro sanitario común
Se aplica a todos los medicamentos veterinarios y productos afines, exceptuando aquellos que estén en el listado armonizado de productos sujetos a registro sanitario simplificado.
Los interesados deben presentar la documentación y cumplir con los requisitos siguientes:
a) Solicitud de registro sanitario (formulario A1 o A2 del Anexo A, según corresponda) debidamente lleno y con los documentos de respaldo correspondientes, firmado y sellado por el regente y el registrante.
b) Poder notariado del titular otorgado a favor del registran te autorizándolo a realizar estas actividades según lo establecido por la autoridad competente de cada Estado Parte.
NOTA 1. Si el poder de representación otorgado previamente se encuentra vigente y faculta al apoderado para registrar, renovar, modificar, cancelar el registro del producto y cualquier otra actividad relacionada con este, no será necesario presentar nuevamente el poder al momento de la solicitud de cualquiera de las anteriores, caso contrario deberá aportarlo.
c) Certificado de libre venta según Anexo B en original, emitido por la autoridad competente del país de origen.
d) Fórmula de composición cuali-cuantitativa completa, en original, emitida por el técnico responsable del laboratorio o por el técnico responsable que designe el fabricante, que incluya el nombre del producto, principios activos y excipientes expresados según el Sistema Internacional de Unidades.
e) Métodos y metodología de análisis físico, químico y biológico, según corresponda, reconocidos internacionalmente o validados por el fabricante para la determinación de la calidad del medicamento o producto afín.
f) Certificado de análisis de un lote comercial del producto terminado, expedido por el fabricante o por el laboratorio autorizado, en original, firmado y sellado por el técnico responsable del mismo. Se aceptará un certificado de análisis de un lote piloto, cuando el producto no ha sido comercializado. Esta disposición no aplica a los productos biológicos.
g) Proyecto de etiquetas, etiquetas, inserto, material de empaque cuando corresponda, para ser aprobados, debiendo cumplir con las disposiciones establecidas por la autoridad competente.
h) Estudios científicos o literatura científica reconocida, que respalden la eficacia, estabilidad, seguridad y calidad, para cada una de las especies solicitadas del producto a registrar, de acuerdo con lo establecido en el Anexo C.
El titular del registro debe aportar un resumen de dichos estudios y sus conclusiones en idioma español (traducción libre) o literatura científica reconocida, según corresponda, señalando las citas bibliográficas y adjuntando la documentación de respaldo completa. Las citas bibliográficas no son requeridas para los medicamentos innovadores.
i) Una muestra del producto a registrar, en el envase original con el que se comercializa en el país de origen, cuando lo requiera la autoridad competente.
j) Estándar analítico (primario o secundario), según lo requiera la autoridad competente, de acuerdo con los programas de control de calidad de cada Estado Parte.
k) Cuando el medicamento veterinario o producto afín sea fabricado, por una empresa distinta al titular del registro sanitario, se debe presentar contrato de maquila actualizado, en original o copia del documento debidamente legalizado, de acuerdo con lo establecido en el Anexo D del presente reglamento.
l) El comprobante de pago por el servicio de registro sanitario cuando corresponda.
m) Para el registro sanitario de los medicamentos veterinarios derivados de la biotecnología, el interesado debe presentar el formulario A1 o A2 del Anexo A, según corresponda, con el producto terminado y la documentación técnica y científica de respaldo indicada en el Formulario para el registro de inmunógenos de subunidades obtenidos por métodos biotecnológicos de CAMEVET, en su versión vigente.
5.3.2 Medicamentos veterinarios con principios activos en combinaciones a dosis fijas
Se exceptúan de este numeral los multivitamínicos, multiminerales o una combinación de éstos, vacunas, kits de diagnóstico, sueros hiperinmunes, productos afines y de medicina alternativa. Si alguno de estos productos se combina con otro tipo de medicamento, debe cumplir con los requisitos de este numeral.
Además de los requisitos para el registro sanitario común, las combinaciones deben cumplir presentando lo siguiente:
5.3.2.1 Ventajas que debe demostrar la combinación. Se aceptan para el registro sanitario las combinaciones de principios activos que, con literatura científica reconocida o estudios científicos específicos para la combinación que se está solicitando, demuestren ventajas farmacológicas significativas de la combinación en relación con cada ingrediente por separado, sin incrementar de manera significativa los riesgos para la salud animal, salud humana y el medio ambiente, ni promover factores de resistencia microbiológica. Por lo tanto, estas combinaciones deben demostrar al menos una de las siguientes ventajas:
a) Efecto sinérgico de sumación o de potenciación: cuando la acción de principios activos diferentes se complementan mejorando la actividad terapéutica.
b) Ampliación del espectro: cuando la combinación de principios activos incrementa el rango de acción contra agentes causales de enfermedades o síndromes ocasionados por varios agentes etiológicos, de manera que la acción de cada ingrediente se complementa para combatir el proceso patológico en su totalidad.
c) Mejor tolerancia y seguridad: cuando la adición de un ingrediente activo disminuye los efectos adversos o colaterales del otro ingrediente o cuando se demuestra que, a dosis inferiores a las normalmente utilizadas para cada ingrediente activo, se obtiene un efecto terapéutico igual o mejor.
d) Mejor efecto clínico-patológico: cuando se demuestre que la adición de un ingrediente activo mejora el efecto clínico-patológico en situaciones específicas, favoreciendo la recuperación del animal.
e) Mejor perfil farmacocinético: cuando la adición de un ingrediente mejora la biodisponibilidad de la formulación, obteniéndose ventajas significativas en la intensidad y duración de la acción.
f) Mejor adaptación a régimen terapéutico poli farmacéutico o facilidad de manejo de la enfermedad.
5.3.2.2 Requisitos que deben cumplir las combinaciones
5.3.2.2.1 Indicaciones de uso. Se aceptan únicamente como válidas las indicaciones en las cuales la combinación de los diferentes principios activos sea necesaria para lograr el efecto terapéutico deseado, de manera que cada ingrediente activo contribuya a lograr tal efecto y estén indicados para lograr tal fin.
5.3.2.2.2 Dosificación. Todos los ingredientes activos deben tener la proporción correcta en la formulación para obtener los efectos deseados, respaldados con estudios realizados que demuestren dicha proporción; con estudios de determinación de dosis y metodología validada.
5.3.2.2.3 Duración de la acción. La duración de la acción de los distintos principios activos combinados debe ser justificada farmacológicamente, de manera que los ingredientes cumplan con todos los usos recomendados.
5.3.2.2.4 Controles de calidad. Debido a que, en las farmacopeas de referencia, la metodología analítica y estándares de calidad son analizados por principios activos individuales, las empresas que deseen registrar estas combinaciones deben presentar los métodos analíticos validados para la identificación y cuantificación de los ingredientes activos presentes en la combinación.
5.3.2.2.5 Antagonismos. Mediante estudios científicos o literatura científica reconocida específicas para la combinación que se está solicitando, debe demostrarse que no existe ningún tipo de antagonismo entre los ingredientes de la formulación.
5.3.2.2.6 Períodos de retiro. Se establece con base en el principio activo que presente el período de retiro más prolongado durante el estudio de eliminación de residuos de la combinación a registrar.
5.3.2.2.7 Vías de aplicación o administración. La vía de administración o aplicación de la combinación debe corresponder con la vía aprobada para cada ingrediente por separado, en caso que se indique una nueva vía deben aportarse los estudios farmacológicos correspondientes o la literatura científica reconocida específica para la combinación que se está solicitando: que demuestre esta nueva vía.
5.4 Registro sanitario simplificado
Se aplica a todos los medicamentos veterinarios y productos afines que se encuentran en el listado de productos sujetos a registro sanitario simplificado. Dicho registro sanitario no implica que estos productos sean de libre venta. Los interesados deben presentar la documentación y cumplir con los siguientes requisitos:
a) Solicitud de registro sanitario simplificado (formulario A3 o A4 del Anexo A, según corresponda).
b) Certificado de libre venta según Anexo B, en original, emitido por la autoridad competente del país de origen, cuando aplique.
c) Certificado de análisis de un lote comercial del producto terminado, expedido por el fabricante o por el laboratorio autorizado, en original, firmado y sellado por el técnico responsable del mismo.
Se aceptará un certificado de análisis de un lote piloto, cuando el producto no ha sido comercializado.
d) Fórmula de composición cuali-cuantitativa completa, en original, emitida por el técnico responsable del laboratorio o por el técnico responsable que designe el fabricante, que incluya el nombre del producto, principios activos y excipientes expresados según el Sistema Internacional de Unidades.
En el caso de los kits de diagnóstico se debe presentar únicamente la fórmula de composición cualitativa completa.
e) Métodos de análisis físico, químico y biológico, según corresponda, utilizados y reconocidos internacionalmente o validados por el fabricante para la determinación de la calidad del medicamento o producto afín.
f) Proyecto de etiquetas, etiquetas, inserto, material de empaque cuando corresponda, para ser aprobados, debiendo cumplir con las disposiciones establecidas por la autoridad competente.
g) Estándar analítico (primario o secundario) según lo requiera la autoridad competente, de acuerdo con los programas de control de calidad de cada Estado Parte.
h) Estudios de estabilidad y estudios de eficacia para el registro de antisépticos y desinfectantes de uso veterinario.
i) Poder notariado del titular otorgado a favor del registrante autorizándolo a realizar estas actividades según lo establecido por la autoridad competente de cada Estado Parte.
NOTA 1. Si el poder de representación otorgado previamente se encuentra vigente y faculta al apoderado para registrar, renovar, modificar, cancelar el registro del producto y cualquier otra actividad relacionada con este, no será necesario presentar nuevamente el poder al momento de la solicitud de cualquiera de las anteriores, caso contrario deberá aportarlo.
j) Una muestra del producto a registrar, en el envase original con el que se comercializa en el país de origen, cuando lo requiera la autoridad competente.
k) Cuando el medicamento veterinario o producto afín sea fabricado, por una empresa distinta al titular del registro sanitario, se debe presentar contrato de maquila actualizado, en original o copia del documento debidamente legalizado, de acuerdo con lo establecido en el Anexo D del presente reglamento.
l) Los literales e), g) y j) no aplican para los kits de diagnóstico de uso veterinario, sin embargo, deben presentar las especificaciones técnicas y componentes del producto terminado.
m) Comprobante de pago por el servicio de registro sanitario cuando corresponda.
5.5 Renovación de registro sanitario
La renovación de registro sanitario se considera una actualización al expediente de registro según la normativa vigente al momento de presentar dicha solicitud, por tanto, adicional a los siguientes puntos, se debe presentar la documentación que se exige en el numeral 5.3.1 y 5.3.2 si corresponde y no fue aportada con anterioridad al expediente:
a) Solicitud de renovación (formulario AS del Anexo A).
b) Documento legal notariado, emitido por el fabricante, el cual debe indicar que las condiciones bajo las que se otorgó el registro sanitario vigente, no han sufrido ninguna modificación legal, técnica ni científica al momento de solicitar la renovación.
c) Certificado de libre venta según Anexo B, en original, emitido por la autoridad competente del país de origen y en caso de productos afines, cuando aplique.
d) Estándar analítico (primario o secundario), según lo requiera la autoridad competente, de acuerdo con los programas de control de calidad de cada Estado Parte.
e) Comprobante de pago por el servicio de registro sanitario cuando corresponda.
f) Poder notariado del titular otorgado a favor del registrante autorizándolo a realizar estas actividades según lo establecido por la autoridad competente de cada Estado Parte.
NOTA 1. Si el poder de representación otorgado previamente se encuentra vigente y faculta al apoderado para registrar, renovar, modificar, cancelar el registro del producto y cualquier otra actividad relacionada con este, no será necesario presentar nuevamente el poder al momento de la solicitud de cualquiera de las anteriores, caso contrario deberá aportarlo.
6. ASPECTOS GENERALES DEL REGISTRO, RENOVACIÓN, MODIFICACIÓN Y RECONOCIMIENTO DE REGISTRO SANITARIOa) Legalización de documentos oficiales. Todos los documentos emitidos por las autoridades oficiales del país de origen del producto, así como poderes especiales que sustentan el registro sanitario, su renovación, modificación o reconocimiento del registro sanitario de medicamentos veterinarios y productos afines, deben cumplir con sus trámites legales y consulares.
b) Los Estados Parte reconocerán una validez máxima de dos años a los Certificados de Libre Venta (CLV), contados a partir de la fecha de su emisión. En el caso de que en el CLV se declaré una validez menor, ésta será la reconocida por los Estados Parte.
La validez del certificado de análisis será de dos años a partir de la fecha de la obtención de resultados finales del análisis.
Ambos deben estar vigentes al momento de la recepción de la solicitud para el registro, renovación, modificación o reconocimiento del registro sanitario.
c) La autoridad competente del Estado Parte podrá registrar medicamentos veterinarios y productos afines elaborados exclusivamente para la exportación, renovar o modificar su registro sanitario, siempre y cuando el establecimiento fabricante cuente con registro y control por la autoridad competente del país de origen; además los productos deberán contar con los controles sanitarios oficiales respectivos y el fundamento científico de que el medicamento no tiene ningún riesgo a la salud humana, animal y ambiente.
d) La autoridad competente de los Estados Parte no registrarán productos que se encuentren en fase experimental en su país de origen.
e) Los medicamentos veterinarios y productos afines se deben registrar con nombre comercial único, no permitiéndose nombres múltiples para un mismo registro sanitario.
f) No se permite que un producto con un mismo número de registro sanitario dirigido para diferentes especies, se comercialice con un material de empaque (etiquetado, imagen comercial) distinto para cada especie, peso o tamaño del animal.
g) Se puede registrar un producto, renovar o modificar su registro sanitario si en su país de origen no ha sido cancelado, prohibido su uso por razones sanitarias, esté en proceso de registro o renovación.
h) La solicitud de renovación debe ser presentada a la autoridad competente desde tres meses antes hasta la fecha de vencimiento del registro sanitario.
Asimismo, se permite la importación y comercialización de productos que se encuentren en proceso de renovación de su registro y en cumplimiento con los requerimientos solicitados por la autoridad competente.
Una vez vencido el registro sanitario, si es de interés del titular del registro mantener su comercialización, debe proceder a tramitar un nuevo registro.
Si durante los tres meses posteriores al vencimiento del registro sanitario de un producto, el interesado solicita se conserve el número de registro sanitario asignado originalmente, para registrarlo posteriormente y presenta justificación, la autoridad competente le mantendrá el número original, sin embargo, durante este periodo, no podrá comercializar el producto. Vencido este plazo, el número quedará inválido.
i) La autoridad competente de los Estados Partes podrá solicitar las certificaciones sanitarias oficiales del país de origen, relacionada con la transmisión de enfermedades o seguridad alimentaria.
j) Los medicamentos veterinarios y productos afines provenientes de Organismos Genéticamente Modificados (OGM) o derivados de la biotecnología, requieren de una evaluación de riesgo; en caso de que los resultados de esta evaluación no sean satisfactorios, será necesario realizar un análisis de riesgo.
k) Los productos biológicos originarios de países con presencia de enfermedades transfronterizas (exóticas), requieren de un análisis de riesgo.
l) La autoridad competente, con la debida justificación técnica, puede solicitar la presentación de documentos adicionales que sustenten la calidad, seguridad o eficacia de los medicamentos veterinarios y productos afines o la aclaración de cualquier información contenida en el expediente de registro sanitario. Si se requiere de controles extras el costo de los mismos debe ser cubierto por el titular del registro.
m) La autoridad competente puede solicitar, supervisar y verificar pruebas de eficacia en las condiciones ambientales del Estado Parte en donde se pretende usar un producto. Igualmente puede ordenar la re-evaluación técnica de las sustancias químicas, agentes biológicos y productos formulados previamente registrados cuando existan indicios de ineficacia, resistencia o de efectos adversos a la salud humana, salud animal o el ambiente.
n) La autoridad competente al comprobar la falsedad de la información de los datos consignados o la alteración de los documentos presentados, denegará o cancelará el registro sanitario según sea el caso.
o) Cuando el medicamento veterinario o producto afín sea fabricado, por una empresa distinta al titular del registro sanitario, se debe presentar contrato de maquila actualizado, en original o copia del documento debidamente legalizado, de acuerdo con lo establecido en el Anexo D del presente reglamento.
p) Para los productos que al momento de renovación de su registro sanitario no cuenten con un estudio de estabilidad acelerado o natural (anaquel), deben presentar para la renovación, el estudio de estabilidad acelerado, o en su defecto, el compromiso del aporte del estudio de estabilidad acelerado.
q) En materia de estudios de estabilidad se tomará la guía armonizada para la elaboración de estudios de estabilidad en productos farmacéuticos veterinarios del Comité Americano de Medicamentos Veterinarios (CAMEVET) en su versión vigente, la cual debe adoptarse por los Estados Parte a través de una Resolución del COMIECO.
r) Hasta que los Estados Parte desarrollen la normativa respectiva en la materia, los estudios de estabilidad indicados en el presente reglamento, relacionados con productos biológicos de uso veterinario, deben cumplir con lo establecido en las normas específicas del país de origen, o en su defecto, con una norma expedida por una entidad reconocida u organismo internacional.
s) Cuando un producto sea elaborado por varios fabricantes, debe contar con un registro sanitario individual por cada uno de los fabricantes, debiendo cumplir con el trámite establecido en el presente reglamento.
t) Los Estados Parte pueden denegar el registro, renovación o reconocimiento del registro sanitario de productos que se destinen a especies productoras de alimentos para el consumo humano que no cuenten con Límite Máximo de Residuos (LMR) establecidos por los organismos internacionales reconocidos en el numeral 17 del presente reglamento.
u) Los Estados Parte pueden prohibir o restringir principios activos de uso veterinario, debiendo publicarlos según los procedimientos internacionales establecidos, cancelar los registros de aquellos productos que contengan sustancias prohibidas que se encuentren vigentes al momento de su entrada en vigor y ordenar el retiro del mercado de los productos prohibidos conforme lo establezca cada Estado Parte.
Asimismo, los titulares de registro de medicamentos veterinarios con registro sanitario vigente, deben modificar las etiquetas para adecuarlas a las restricciones aprobadas conforme lo establezca cada Estado Parte.
7. EXENCIÓN DE REGISTRO SANITARIO
Se eximen del registro sanitario los productos en los siguientes casos:
a) Ante una emergencia decretada, eventualidad sanitaria o en protección a un interés público nacional.
b) Con fines de investigación, previo cumplimiento de los requisitos establecidos en el Anexo F y de acuerdo con el protocolo de ensayo clínico establecido en el Anexo L del presente reglamento, según corresponda.
c) Para campañas sanitarias oficiales.
d) Muestras médicas con fines de registro sanitario.
e) Donaciones.
f) Medicamentos veterinarios bajo prescripción veterinaria, destinados al tratamiento de un animal específico cuando no exista ningún producto similar registrado en el Estado Parte.
No se permite la solicitud de exención de registro sanitario para principios activos prohibidos, para enfermedades de notificación transfronteriza (exóticas) o que se encuentren restringidos para las especies a las que se destine el producto, según cada Estado Parte.
Las autoridades competentes deben analizar las solicitudes de exención de registro, valorando que las mismas cumplan con lo establecido en el presente reglamento y que el producto no constituya riesgo inadmisible a la salud pública, salud animal o al ambiente, debiendo las autoridades determinar la cantidad, fines y los requisitos a cumplir según lo establecido por cada Estado Parte.8. NÚMERO Y CERTIFICADO DE REGISTRO SANITARIO
En el caso de que la autoridad competente apruebe el registro sanitario, inscribirá el producto según corresponda, asignándole un número con el cual se identificará el producto de uso veterinario de acuerdo con la codificación armonizada.
Una vez inscrito el medicamento veterinario o producto afín, la autoridad competente extenderá el certificado de registro sanitario oficial correspondiente.
9. PLAZO DEL REGISTRO SANITARIO
El registro sanitario concedido tiene una validez de cinco (5) años a partir de su inscripción, pudiendo ser renovado por períodos iguales a solicitud del interesado. Sin embargo, cuando se infrinja lo estipulado en este reglamento o se demuestre que las condiciones originales del registro sanitario han variado en cuanto a eficacia, indicaciones o seguridad del producto, se procederá a exigir las correcciones necesarias o la anulación del registro sanitario.
10. CONDICIONES PARA LA IMPORTACIÓN
La autoridad competente del Estado Parte solamente autorizará solicitudes de importación con fines comerciales, de productos que cuenten con el registro sanitario debidamente aprobado y vigente o en proceso de renovación y aquellos casos contemplados en el numeral 7 (exenciones del registro sanitario).
11. ARCHIVO O DESESTIMACIÓN DE LA GESTIÓN
Se debe ordenar el archivo o desestimación de la gestión, según los procedimientos administrativos de cada Estado Parte, cuando las solicitudes presentadas no reúnan los requisitos señalados en el presente reglamento, la información presentada se encuentre incompleta o inconforme para los fines requeridos o por haber sido presentada en forma extemporánea, siempre que hubiera sido debidamente prevenida o requerida por la autoridad competente.
12. ANOTACIÓN MARGINAL O MODIFICACIÓN DEL REGISTRO SANITARIO
El registro sanitario de un producto puede ser modificado a petición del registrante y del regente, previo pago del valor cuando corresponda, únicamente para los casos señalados en el Anexo E "Tipología y Requisitos de Modificación de Registro Sanitario". Para ello debe aportar el Poder notariado del titular otorgado a favor del registrante autorizándolo a realizar estas actividades según lo establecido por la autoridad competente de cada Estado Parte.
NOTA 1. Si el poder de representación otorgado previamente se encuentra vigente y faculta al apoderado para registrar, renovar, modificar, cancelar el registro del producto y cualquier otra actividad relacionada con este, no será necesario presentar nuevamente el poder al momento de la solicitud de cualquiera de las anteriores, caso contrario deberá aportarlo.
Para los casos no contemplados en el Anexo E, se requiere de un registro sanitario distinto e individual para cada una de las variaciones. Las anotaciones marginales o modificaciones deben ser resueltas (aprobadas o denegadas) y notificadas al interesado por escrito por parte de la autoridad competente. No implican un nuevo registro sanitario, siempre que se encuentren vigentes. En caso de no ser aprobadas, se deben indicar las razones de la denegación.
13. CANCELACIÓN DE REGISTRO
El registro sanitario de cualquier producto podrá ser cancelado por la autoridad competente, previo a su vencimiento, cuando:
a) Lo solicite por escrito el propietario del registro sanitario.
b) No cumpla en tres muestreos consecutivos de tres lotes diferentes con las normas de calidad establecidas en su registro sanitario para dicho medicamento veterinario o producto afín.
c) El uso y manipulación del producto represente riesgo inadmisible comprobado para la salud humana, salud animal o el ambiente.
d) Se detecte irregularidad, fraude o falsedad en la composición del producto o en la información aportada para su registro sanitario.
e) Se compruebe, mediante estudios o ensayos reconocidos por la autoridad competente del Estado Parte, que el producto es ineficaz para los fines indicados en el registro sanitario.
f) Alguno de los componentes del producto sea prohibido por el Estado Parte.
g) Se compruebe que su uso interfiere con los objetivos de los programas nacionales de vigilancia epidemiológica, la erradicación de enfermedades, la preservación o mejora del estatus sanitario.
14. REQUISITOS DE ETIQUETADO Y RE-ETIQUETADO
El proyecto de etiqueta o etiqueta, estuche e inserto que deben acompañar a la solicitud de registro sanitario deben ser presentados en español. Adicionalmente, a petición del interesado, se puede incluir otro idioma.
14.1 Obligatoriedad de la etiqueta y re-etiquetado
Todo medicamento veterinario o producto afín que se fabrique, manipule, almacene, fraccione, distribuya, importe, comercialice, despache, expenda o utilice en los países de la región centroamericana, debe contener la respectiva etiqueta que cumpla con lo estipulado en el presente reglamento.
Debe tener un tamaño de letra legible a simple vista, no menor a 1,5 mm (4 puntos Didot), llevar claramente impresa, visible y en tinta indeleble la información en nomenclatura internacionalmente aceptada, expresando las unidades de acuerdo con el Sistema Internacional de Unidades, no desprenderse y mantener la calidad de las especificaciones definidas por el fabricante.
Se permite el uso de tinta indeleble (jet printer) para el número de registro, fecha de fabricación, fecha de vencimiento y número de lote.
14.2 Contenido de etiqueta común
a) Nombre del producto.
b) Forma farmacéutica.
c) Vía de administración o aplicación.
d) Principios activos/ agente biológico y su concentración.
e) Contenido neto.
f) Nombre y país del laboratorio fabricante. En caso de fabricación a terceros, debe estar especificado (elaborado por ... para ... ).
g) Número de lote, fecha de fabricación y fecha de vencimiento, expresado en mm/aa (mes/año).
h) Fecha de caducidad o su pictograma una vez abierto el producto, cuando aplique.
i) Requisitos para el almacenamiento y conservación.
j) Número de registro sanitario del Estado Parte donde se registra, puede ser impreso en el estuche (caja) si la contiene.
k) La frase "Venta bajo receta médica" (Para medicamentos controlados)
l) La frase "Uso veterinario".
m) Especie de destino detallando cada una de ellas.
n) El pictograma de la especie animal a que se destina (opcional).
o) La frase "Lea el inserto antes de utilizar el producto", cuando lo contiene.
Esta información debe venir impresa desde el país de origen.
No se permite el re-etiquetado, ni el uso de etiquetas auto adhesibles o auto adheribles ("stickers") para ninguna información en las etiquetas finales, excepto para el número de registro sanitario, previa aprobación de la autoridad competente.
14.3 Contenido de etiqueta para productos de higiene y belleza
a) Nombre del producto.
b) Forma cosmética.
c) Instrucciones de uso.
d) Ingredientes (de mayor a menor concentración).
e) Contenido neto.
f) Nombre y país del laboratorio fabricante.
En caso de fabricación a terceros, debe estar especificado (elaborado por ... para ...).
g) Número de lote, fecha de fabricación y fecha de vencimiento, expresado en mm/aa (mes/año).
h) Número de registro sanitario del Estado Parte donde se registra.
i) La frase "Uso veterinario".
j) Especie de destino detallando cada una de ellas.
k) El pictograma de la especie animal a que se destina (opcional).
l) La frase "Lea el inserto antes de utilizar el producto", cuando lo contiene.
En este tipo de productos se permite el re-etiquetado, sin que se oculte el número de lote, fecha de fabricación, fecha de vencimiento, fabricante y país de origen; esta información debe venir impresa en la etiqueta o envase desde el país de origen.
14.4 Contenido de etiqueta para kits de diagnóstico.
a) Nombre del producto.
b) Instrucciones de uso. Agregar la frase "Lea el inserto antes de utilizar el producto".
c) Componentes y composición.
d) Contenido neto.
e) Nombre y país del laboratorio fabricante. En caso de fabricación a terceros, debe estar especificado (elaborado por ... para ...).
f) Número de lote, fecha de fabricación y fecha de vencimiento, expresado en mm/aa (mes/año).
g) Número de registro sanitario del Estado Parte donde se registra.
h) La frase "Uso veterinario".
i) Especie de interés diagnóstico, detallando cada una de ellas.
j) El pictograma de la especie animal a que se destina (opcional).
k) Condiciones de almacenamiento.
En este tipo de productos se permite el re-etiquetado, sin que se oculte el número de lote, fecha de fabricación, fecha de vencimiento, fabricante y país de origen; esta información debe venir impresa en la etiqueta o envase desde el país de origen.
14.5 Contenido de etiqueta para envases o empaques menores o iguales a cincuenta mililitros, cien gramos, ampollas colapsibles y blíster
Las etiquetas de envases o empaques menores o iguales a cincuenta (50) mililitros, cien (100) gramos, ampollas colapsibles y blíster, en su envase o empaque primario deben indicar al menos la siguiente información:
a) Nombre del producto.
b) Contenido neto, no aplica para blíster.
c) Condiciones de almacenamiento, no aplica para blíster.
d) Principios activos y su concentración.
e) Número de lote y fecha de vencimiento, expresado en mm/aa (mes/año).
f) Nombre del país y laboratorio fabricante. En caso de fabricación a terceros, debe estar especificado (elaborado por ... para ...).
g) La frase "Uso veterinario".
Esta información debe venir impresa en la etiqueta, envase o empaque desde el país de origen.
La información faltante según numeral 14.2 y el número de registro sanitario, deben estar contenidas en el empaque secundario (caja) si la contiene y el inserto adjunto.
Cada unidad de venta al público debe acompañarse del correspondiente inserto.
14.6 Contenido de etiqueta para envases o empaques menores o iguales a cincuenta mililitros y cien gramos para ectoparasiticidas de uso veterinario
Las etiquetas de envases o empaques menores o iguales a cincuenta (50) mililitros y cien (100) gramos, en su envase o empaque primario deben indicar al menos la siguiente información:
a) Nombre del producto.
b) Contenido neto.
c) Condiciones de almacenamiento
d) Principios activos y su concentración.
e) Número de lote y fecha de vencimiento, expresado en mm/aa (mes/año).
f) Nombre del país y laboratorio fabricante. En caso de fabricación a terceros, debe estar especificado (elaborado por ... para ... ).
g) La frase "Uso veterinario".
h) Período de retiro, cuando aplique.
i) Clase y tipo de ectoparasiticida.
j) Antídoto(s) en caso de intoxicaciones en seres humanos.
k) Instrucciones de uso. Lea el inserto antes de utilizar el producto, cuando aplique.
Esta información debe venir impresa en la etiqueta, envase o empaque desde el país de origen.
La información faltante según numeral 14.2 y el número de registro sanitario, deben estar contenidas en el empaque secundario (caja) si la contiene o el inserto adjunto.
Cada unidad de venta al público debe acompañarse del correspondiente inserto.
14.7 Contenido del envase o empaque secundario
El envase o empaque secundario (caja - estuche) debe contener la siguiente información:
a) Nombre del producto.
b) Forma farmacéutica.
c) Vía de administración o aplicación.
d) Principios activos/ agente biológico y su concentración.
e) Contenido neto.
f) Nombre y país del laboratorio fabricante. En caso de fabricación a terceros, debe estar especificado (elaborado por ... para ...).
g) Número de lote, fecha de fabricación y fecha de vencimiento, expresado en mm/aa (mes/año).
h) Fecha de caducidad o su pictograma una vez abierto el producto, cuando aplique.
i) Requisitos para el almacenamiento y conservación.
j) Número de registro sanitario del Estado Parte donde se registra.
k) La frase "Venta bajo receta médica" (Para medicamentos controlados).
l) La frase "Uso veterinario".
m) El pictograma de la especie animal(es) a que se destina (opcional).
n) Especie de destino detallando cada una de ellas.
o) La frase "Lea el inserto antes de utilizar el producto", cuando lo contiene.
p) Clase farmacológica.
q) Indicaciones.
r) Contraindicaciones y restricciones.
s) Dosis por especie animal.
t) Advertencia y precauciones.
u) Período de retiro, cuando aplique.
v) La frase "Conservar fuera del alcance de los niños y animales domésticos".
Si la presentación del producto no contiene empaque secundario, la totalidad de la información requerida en este numeral, debe ser impresa en la etiqueta de envase o empaque primario. Esta información debe venir impresa desde el país de origen.
14.8 Contenido del inserto
Cuando se requiera de inserto, éste debe acompañar siempre al producto al ser distribuido ya sea al por mayor o al detalle, incluido en el envase o empaque secundario (caja-estuche) o adherido al producto si no contiene caja.
Debe contener la siguiente información:
a) Nombre del producto.
b) Forma farmacéutica.
c). Vía de administración o aplicación.
d) Principios activos/ agente biológico y su concentración.
e) Presentaciones (opcional).
f) Nombre y país del laboratorio fabricante. En caso de fabricación a terceros, debe estar especificado (elaborado por ...... para ...).
g) Requisitos para el almacenamiento y conservación.
h) Número de registro sanitario del Estado Parte donde se registra (opcional).
i) Agregar la frase "Venta bajo receta médica" (Para medicamentos controlados).
j) Clase farmacológica.
k) Indicaciones.
l) Especie de destino detallando cada una de ellas.
m) Dosis por especie animal.
n) Advertencia y precauciones.
o) Período de retiro, si aplica.
p) Agregar la frase "Conservar fuera del alcance de los niños y animales domésticos".
q) Contraindicaciones y restricciones.
r) Efectos colaterales.
s) Reacciones adversas.
t) Antídotos, si existen.
14.9 Contenido del inserto para kits de diagnóstico
Cuando se requiera de inserto, éste debe acompañar siempre al producto al ser distribuido ya sea al por mayor o al detalle, incluido en el envase o empaque secundario (caja-estuche) o adherido al producto si no contiene caja.
Debe contener la siguiente información:
a) Nombre del producto.
b) Instrucciones de uso.
c) Componentes y composición.
d) Contenido neto.
e) Especificaciones técnicas del producto terminado.
f) Nombre y país del laboratorio fabricante. En caso de fabricación a terceros, debe estar especificado (elaborado por ... para ...).
g) Número de registro sanitario del Estado Parte donde se registra (opcional).
h) Agregar La frase "Uso veterinario".
i) Especie de interés diagnóstico.
j) El pictograma de la especie animal a que se destina (opcional).
k) Requisitos para el almacenamiento y conservación.
l) Advertencias y precauciones.
m) Agregar la frase "Conservar fuera del alcance de los niños y animales domésticos".
14.10 Contenido de etiqueta, estuche e inserto para ectoparasiticidas de uso veterinario
Debe incluir, además:
a) Clase y tipo de ectoparasiticida.
b) Condiciones de uso adecuado concordante con lo declarado en la solicitud de registro sanitario, especificando los nombres comunes y científicos de las plagas o parásitos a combatir, así como el modo de utilización y de aplicación en los animales, según la plaga de que se trate.
c) Método de preparar el material final de aplicaciones, cuando proceda.
d) Métodos para la descontaminación y disposición final de envases usados, derrames permanentes y ectoparasiticidas no utilizados.
e) Advertencias y precauciones para el uso, relativos a la toxicidad de los ingredientes para seres humanos y animales, síntomas de intoxicación, primeros auxilios y medidas aplicables en caso de intoxicación oral, dérmica o inhalatoria cuando proceda, antídoto(s) e indicaciones para el tratamiento.
f) En mayúscula, en negrita y en un color contrastante, la leyenda: "EN CASO DE INTOXICACIÓN CONSULTE AL MÉDICO Y ENTRÉGUELE ESTA ETIQUETA".
g) La leyenda destacada "Manténgase alejado de los niños, animales y alimentos".
h) La leyenda destacada que diga: "ALTO: Lea esta etiqueta antes de usar el producto".
i) Indicaciones sobre medidas de protección al medio ambiente.
j) Indicaciones del equipo de protección recomendado para la aplicación del producto, cuando aplique.
k) El número de teléfono del Centro Nacional de Intoxicaciones del país donde se registra.
La clasificación toxicológica del ectoparasiticida debe realizarse según la Organización Mundial de la Salud (OMS), debe presentarse en el etiquetado de manera visual mediante un color especifico y su identificación se debe hacer mediante una banda a lo largo de la base de la etiqueta y estuche secundario, si lo contiene, cuyo ancho será no menor al 15% de la altura de dicha etiqueta y estuche.
Al centro de la banda debe imprimirse en letras de color negro o en un color contrastante el texto que señala la categoría toxicológica del producto "EXTREMADAMENTE PELIGROSO", "ALTAMENTE PELIGROSO'', "MODERADAMENTE PELIGROSO" o "LIGERAMENTE PELIGROSO" según corresponda, en un tamaño no menor de la tercera parte del ancho de la banda. En la etiqueta y estuche secundario, si lo contiene, deben colocarse pictogramas ilustrativos que apoyen el uso adecuado del producto en un tamaño que no exceda de las dos terceras partes del ancho.
Las tonalidades de los colores (pantones) así como los símbolos y palabras de advertencia para identificar la categoría toxicológica de los ectoparasiticidas de uso veterinario, se debe hacer de acuerdo con la clasificación del International Programme on Chemical Safety (IPCS) de la Organización Mundial de la Salud (OMS).
La determinación de la banda toxicológica para ectoparasiticidas con un ingrediente activo o combinaciones se debe realizar de acuerdo con las fórmulas recomendadas por el IPCS. De igual forma se deben utilizar las DLSO recomendadas por dicho programa.
La superficie total de la etiqueta puede ser de otros colores, excepto la franja correspondiente a la categoría toxicológica. El contraste entre el texto impreso y el fondo debe resaltar.
la legibilidad de los caracteres y no interferir con el color de la franja.
Para las ampollas colapsibles y blíster, el contenido de la etiqueta, envase o empaque primario se debe regir según se indica en el numeral 14.5 de este reglamento. Para presentaciones menores o iguales a cincuenta (50) mi o cien (100) gramos, el contenido de la etiqueta, envase o empaque primario se debe regir según se indica en el numeral 14.6 de este reglamento.
14.11 Etiquetas de muestras médicas
Todo medicamento veterinario o producto afín catalogado como "muestra médica", debe tener las leyendas "MUESTRA MÉDICA VETERINARIA" "PROHIBIDA SU VENTA". La información incluida debe contener al menos la lista completa de ingredientes activos, forma de administración adecuada y contraindicaciones. Queda prohibida la venta y comercio de muestras de medicamentos veterinarios y productos afines, así como la exposición de éstos en los establecimientos que los comercialicen.15. CONTROL DE ESTABLECIMIENTOS Y DE LOS MEDICAMENTOS VETERINARIOS Y PRODUCTOS AFINES
15.1 Los países miembros, deben establecer las medidas y actividades de control y fiscalización sobre los establecimientos y productos involucrados en el registro sanitario, la fabricación, fraccionamiento, importación, almacenamiento, exportación, reexportación, reempaque, distribución, despacho, expendio, manejo y uso de medicamentos veterinarios y productos afines.
15.2 De la publicidad y sus prohibiciones
La publicidad para los productos del grupo III y IV en cualquier medio de comunicación, no debe contener ambigüedades, omisiones o exageraciones que entrañen la posibilidad de inducir a error al usuario, en particular, en lo que respecta a la seguridad sobre el uso, manejo, naturaleza y composición del producto de uso veterinario. No puede contener información diferente de la que ampara el registro sanitario del producto.
La publicidad se debe hacer de acuerdo con la información técnica de eficacia comprobada por los estudios correspondientes en su registro sanitario.
Se prohíbe toda publicidad de los medicamentos veterinarios del grupo I y II.
Se prohíbe la publicidad o propaganda de aquellos productos que no se encuentren registrados y el uso de imágenes que lesionen la dignidad humana.
16. CONFIDENCIALIDAD Y SEGURIDAD DE LA INFORMACIÓN
El personal de las entidades responsables del registro sanitario, debe cumplir con lo señalado por los instrumentos. jurídicos de cada Estado Parte respecto a la confidencialidad y seguridad en el manejo de la información objeto de trámite, debiendo guardar secreto administrativo sobre la documentación que así lo requiera en el desempeño de sus funciones.
La información técnica aportada para el registro sanitario, considerada confidencial, podrá ser usada por la autoridad competente con fines de preservación de la salud pública, salud animal y ambiente, según lo que señalen las leyes y normas locales respectivas.
17. METODOLOGÍAS ANALÍTICAS Y ESPECIFICACIONES DE CALIDAD
La autoridad competente aplicará las guías de referencia de metodologías analíticas, especificaciones de inocuidad, control y calidad en medicamentos veterinarios y productos afines contemplados en:
1. Codex Alimentarius.
2. Código de Regulaciones Federales (CFR) de los Estados Unidos de América, Títulos 9 y 21.
3. Food Safety and Inspection Service (SFIS), USDA.
4. Organización Mundial de Sanidad Animal (O.LE.).
5. Asociación Oficial de Químicos Analíticos (AOAC).
6. Farmacopea de los Estados Unidos de América (USP).
7. Farmacopea de la Unión Europea.
8. Reglamento Técnico Centroamericano RTCA 11.03.39:06 Productos Farmacéuticos. Reglamento de Validación de Métodos Analíticos para la evaluación de la Calidad de los Medicamentos o en su versión vigente.
9. Environmental Protection Agency (EPA).
10. Metodologías de análisis validadas por el fabricante de acuerdo con las guías de referencia reconocidas.
Los Estados Parte reconocen los Límites Máximos de Residuos (LMR) de medicamentos veterinarios establecidos por:
1. Codex Alimentarius.
2. Food and Drugs Administration (FDA).
3. Agencia Europea del Medicamento (EMA por sus siglas en inglés).
4. Comité Mixto FAO/OMS de Expertos en Aditivos Alimentarios (JECFA por sus siglas en inglés).
5. Otros entes gubernamentales y/o de investigación de reconocimiento internacional.
18. VIGILANCIA Y VERIFICACIÓN
Corresponde la vigilancia y verificación del presente reglamento a las autoridades competentes de los Estados Parte.
19. BIBLIOGRAFÍA1. Comité para las Américas de Medicamentos Veterinarios (CAMEVET–OIE), Documentos aprobados.
2. CDM Compendium Canadiense de Medicamentos.
3. FAO, Directrices en materia de medicamentos veterinarios y productos afines.
4. Legislación vigente de cada Estado Parte en materia de medicamentos veterinarios y productos afines.
5. OIRSA, Requisitos Técnico Administrativos para el Registro sanitario y Control de Medicamentos veterinarios y productos afines y Alimentos Para Animales.
6. Real Decreto 1246-2008 de Medicamentos Veterinarios.
20. REFERENCIAS
1. Código Federal de Regulaciones (CFR) del Food and Drugs Administration (FDA), títulos 9 y 21.
2. Food Safety and Inspection Service (SFIS), USDA.
3. Organización Mundial de la Salud (OMS).
4. Codex Alimentarius, Límites Máximos de Residuos (LMR) aprobados para medicamentos y ectoparasiticidas de uso veterinario.
5. Agencia Europea del Medicamento, Límites Máximos de Residuos (LMR) aprobados para medicamentos y ectoparasiticidas de uso veterinario.
6. United States Pharmacopoeia.
7. Farmacopea Europea. 8. International Conference of Harmonization (ICH).
9. Veterinary International Conference of Harmonization (VICH).
10. Organización Mundial de Sanidad Animal (OIE).
11. Organización para la Cooperación y el Desarrollo Económico (OCDE).
ANEXO A
(NORMATIVO)
FORMULARIO DE SOLICITUD DE REGISTRO SANITARIO
A1- FORMULARIO DE SOLICITUD DE REGISTRO SANITARIO PARA MEDICAMENTOS, QUÍMICOS Y ECTOPARASITICIDAS DE USO VETERINARIO
1. NOMBRE COMERCIAL DEL PRODUCTO
2. CLASIFICACIÓN (uso oficial exclusivo)
3. ESTABLECIMIENTO SOLICITANTE: PROPIETARIO /REPRESENTANTE LEGAL
3.1. Nombre.
3.2. Dirección exacta.
3.3. País.
3.4. Número de registro sanitario del establecimiento registrante.
3.5. Responsable técnico.
3.5.1. Número de identificación profesional.
3.5.2. Lugar o medio para recibir notificaciones.
4. ESTABLECIMIENTO ELABORADOR
4.1. Nombre.
4.2. Dirección exacta.
4.3. País.
4.4. Número de registro sanitario del establecimiento.
5. ESTABLECIMIENTO FRACCIONADOR
5.1. Nombre.
5.2. Dirección exacta.
5.3. País.
5.4. Número de registro sanitario del establecimiento.
6. FORMA FARMACÉUTICA
7. ESPECIFICACIONES Y MÉTODOS DE CONTROL DE LOS COMPONENTES DE LA FÓRMULA.
8. METODOLOGÍA DE ELABORACIÓN DEL PRODUCTO. (Describir resumidamente el proceso de fabricación).
9. ESPECIFICACIONES Y MÉTODOS DE CONTROL DEL PRODUCTO TERMINADO
Indicar y describir las especificaciones y los métodos que se utilizan en la evaluación cuali-cuantitativa de los componentes de la formulación en el producto terminado.
9.1. Métodos Biológicos
9.2. Métodos Microbiológicos.
9.3. Métodos Químicos
9.4. Métodos Físicos
9.5. Métodos Físico-químicos
10. FORMA DE PRESENTACIÓN Y CANTIDAD DE PRODUCTO
11. ESPECIFICACIÓN Y CONTROL DE ENVASES
11. 1. Características del envase
11.2. Sistema de inviolabilidad
11.3. Control de calidad de envases
12. PERÍODO DE VALIDEZ (Vencimiento del lote)
13. PRUEBAS DE EFICACIA (Antecedentes bibliográficos y/o pruebas clínicas de eficacia, cuando corresponda).
14. INDICACIONES DE USO Y CATEGORÍA DE COMERCIALIZACIÓN
14.1. Principales o complementarias.
14.2. Para productos antimicrobianos y antiparasitarios, especificar los agentes etiológicos susceptibles.
14.3. Especies animales y categorías a las que se destina, uso específico en instalaciones, equipos, u otros.
14.4. Categorización oficial: (libre venta, venta bajo receta médica, controlado y restringido u otros tipos de venta).
15. VÍA DE APLICACIÓN Y FORMA DE ADMINISTRACIÓN O UTILIZACIÓN DEL PRODUCTO Parenteral, oral, instalaciones, equipos, instrumentales u otras.
16. PRODUCTOS DE PREPARACIÓN EXTEMPORÁNEA
16.1. Preparación del producto para su correcto uso (Premezclas, soluciones, pre-emulsiones, suspensiones u otras).
16.2. Período de validez después de su reconstitución, avalada por estudios de estabilidad.
16.3. Cuando el producto sea para administrarse en raciones o en el agua de bebida debe indicarse su estabilidad, compatibilidad y/o tiempo de permanencia eficaz en la mezcla o en la solución.
17. DOSIFICACIÓN
Indicar la o las cantidades del o de los principios activos expresadas en unidades de peso, volumen o Unidades Internacionales (UI) por kg de peso vivo de acuerdo con su indicación de uso para las diferentes especies y edades.
17.1. Dosis del producto de acuerdo con su indicación de uso por peso vivo según especies y edad.
17.2. Intervalo entre dosis.
17.3. Duración del tratamiento.
18. ESTUDIOS DE SEGURIDAD
Deben incluirse los antecedentes bibliográficos de seguridad e inocuidad o los estudios clínicos que respaldan estos aspectos, según corresponda.
19. FARMACOCINÉTICA DEL PRODUCTO
Vías de absorción, distribución y eliminación de los principios activos o sus metabólicos.
20. FARMACODINAMIA DEL PRODUCTO (Resumen)
21. EFECTOS COLATERALES POSIBLES (Locales o generales), INCOMPATIBILIDADES Y ANTAGONISMOS FARMACOLÓGICOS
21.1. Contraindicaciones y limitaciones de uso (casos en que su administración puede dar lugar a efectos nocivos).
21.2. Precauciones que deben adoptarse antes, durante o después de su administración.
22. INTOXICACIÓN Y SOBREDOSIS EN LOS ANIMALES
22.1 Margen de seguridad e inocuidad en la especie diana o destino.
22.2 Síntomas, conducta de emergencia y antídotos.
23. INTOXICACIÓN EN EL HOMBRE
23.1 Categoría toxicológica.
23.2 Se debe indicar tratamiento, antídoto y datos de centros toxicológicos de referencia en el país.
24. EFECTOS BIOLÓGICOS NO DESEADOS.
Se debe declarar si el o los componentes activos en las condiciones indicadas de uso, producen efectos adversos como los que a continuación se mencionan, debiéndose aportar, si existiera, la bibliografía científica al respecto.
24.1. Carcinogénesis
24.2. Teratogénesis.
24.3. Mutagénesis
24.4. Resistencia a agentes patógenos
24.5. Discrasias sanguíneas
24.6. Neurotoxicidad
24. 7. Hipersensibilidad
24.8. Sobre la reproducción 24.9. Sobre la flora normal
25. CONTROLES SOBRE RESIDUOS MEDICAMENTOSOS
25.1. Datos sobre Ingesta Diaria Admisible {IDA)
25.2. Límite Máximo de Residuos (LMR) en tejidos (músculo, hígado, riñón, grasa), leche, huevos y miel.
25.3. Tiempo que debe transcurrir entre el último tratamiento y el sacrificio del animal para consumo humano.
25.4. Tiempo que debe transcurrir entre el último tratamiento y el destino de la leche, huevos o miel para consumo humano.
26. PRECAUCIONES GENERALES
26.1. Límite máximo y mínimo de temperatura para su correcta conservación.
26.2. Describir la forma adecuada de almacenamiento, transporte y destrucción del producto, así como también del método de disposición final de los envases que constituyan un factor de riesgo para la salud pública, animal y el medio ambiente.
27. CAUSAS QUE PUEDEN HACER VARIAR LA CALIDAD DEL PRODUCTO Precipitaciones, disociaciones, disminución o pérdida de actividad de los principios activos, frío, calor, luz, humedad, compresión en estibas o depósitos.
28. TRABAJOS CIENTÍFICOS O MONOGRAFÍAS. Se deben adjuntar los trabajos científicos o monografías relacionadas con el producto. Se debe incluir la traducción del resumen y las conclusiones de dichos trabajos en idioma español.
Se adjuntan a esta solicitud los requisitos de registro establecidos en este reglamento.
Toda la información que se adjunta a esta solicitud, es parte integral de la misma.
Declaramos que la información presentada es verdadera y toda alteración o información falsa, invalida esta solicitud, sin menoscabo de la responsabilidad penal que ello implica.
Nombre y firma del regente _____________
Lugar y Fecha ______
Nombre y firma del registrante_________
A2- FORMULARIO DE SOLICITUD DE REGISTRO SANITARIO PARA PRODUCTOS BIOLÓGICOS DE USO VETERINARIO
1. NOMBRE COMERCIAL DEL PRODUCTO
2. NOMBRE COMÚN Y CLASIFICACIÓN - (uso oficial exclusivo).
3. ESTABLECIMIENTO SOLICITANTE: PROPIETARIO /REPRESENTANTE LEGAL
3.1. Nombre.
3.2. Dirección exacta.
3.3. País.
3.4. Número de registro sanitario del establecimiento registrante.
3.5. Responsable técnico.
3.5.1. Número de identificación profesional.
.
3.5.2. Lugar o medio para recibir notificaciones.
4. ESTABLECIMIENTO ELABORADOR
4.1. Nombre.
4.2. Dirección exacta.
4.3. País.
4.4. Número de registro sanitario del establecimiento.
5. ESTABLECIMIENTO FRACCIONADOR
5.1. Nombre.
5.2. Dirección exacta.
5.3. País.
5.4. Número de registro sanitario del establecimiento.
6. DEFINICIÓN DE LINEA BIOLÓGICA (Antígenos vacunales, sueros hiperinmunes, reactivos para diagnóstico)
7. FORMA FARMACÉUTICA
8. FORMULA CUALI-CUANTITATIVA -CONSTITUCIÓN BIOLÓGICA Y QUÍMICA
8.1. Antígeno: identificación, cantidad por dosis (título, masa antigénica, proteína u otros)
8.2. Sueros hiperinmunes: concentración en U.I.
8.3. Inactivantes; conservadores; estabilizadores; emulsificantes, adyuvantes u otras sustancias.
8.4. Diluyente: constitución química y biológica si la contiene.
9. METODOLOGÍA DE ELABORACIÓN DEL PRODUCTO
9.1. Describir resumidamente el proceso de fabricación. Describa todos los pasos necesarios para el desarrollo del formulario de inscripción.
9.2. Métodos de control del producto en proceso.
10. FORMA DE PRESENTACIÓN Y CONTENIDO
11. ESPECIFICACIÓN Y CONTROL DE ENVASES
11.1. Características del envase.
11.2. Sistema de inviolabilidad.
11.3. Control de calidad de envases.
12. PERÍODO DE VALIDEZ (Vencimiento)
13. INDICACIONES DE USO Y CATEGORÍA DE COMERCIALIZACIÓN
13.1. Indicaciones principales o complementarias.
13.2. Especies animales a las que se destina.
14. VÍA DE APLICACIÓN Y FORMA DE ADMINISTRACIÓN OUTILIZACIÓN DEL PRODUCTO (Parenteral, oral, dérmica, pulverización, escarificación, ocular, nasal u otras).
15. PRODUCTOS DE PREPARACIÓN EXTEMPORÁNEA
15.1. Preparación del producto para su correcto uso.
15.2. Indicar período de validez después de su reconstitución avalada por estudios de estabilidad.
16. DOSIFICACIÓN
16.1. Dosis del producto en aplicación preventiva o curativa por peso vivo según especies y edad.
16.2. Esquema de aplicación recomendado.
16.3. Tiempo necesario para conferir inmunidad y duración de la misma.
17. EFECTOS COLATERALES POSIBLES (Locales o generales), INCOMPATIBILIDADES Y ANTAGONISMOS
17.1. Contraindicaciones y limitaciones de uso (casos en que su administración pueda dar lugar a efectos nocivos).
17.2. Precauciones que deben adoptarse antes, durante o después de su administración.
18. PRECAUCIONES GENERALES
Límite máximo y mínimo de temperatura para su correcta conservación. Describir la forma adecuada de almacenamiento, transporte y destrucción del producto, así como también del método de eliminación de los envases que constituyan un factor de riesgo para la salud pública, animal y el medio ambiente.
Se adjuntan a esta solicitud los requisitos de registro establecidos en este reglamento.
Toda la información que se adjunta a esta solicitud, es parte integral de la misma.
Declaramos que la información presentada es verdadera y toda alteración o información falsa, invalida esta solicitud, sin menoscabo de la responsabilidad penal que ello implica.
Nombre y firma del regente____________
Lugar y Fecha ______
Nombre y firma del registrante__________
A3- FORMULARIO DE SOLICITUD DE REGISTRO SANITARIO SIMPLIFICADO. A EXCEPCIÓN DE KIT DE DIAGNÓSTICO DE ENFERMEDADES
1. NOMBRE COMERCIAL DEL PRODUCTO
2. CLASIFICACIÓN (uso oficial exclusivo)
3. ESTABLECIMIENTO SOLICITANTE: PROPIETARIO/REPRESENTANTE LEGAL
3.1. Nombre
3.2. Dirección exacta
3.3. País
3.4. Número de registro sanitario del establecimiento registrante
3.5. Responsable técnico
3.5.1. Número de identificación profesional
3.5.2. Lugar o medio para recibir notificaciones
4. ESTABLECIMIENTO ELABORADOR
4.1. Nombre
4.2. Dirección exacta
4.3. País
4.4. Número de registro sanitario del establecimiento
5. ESTABLECIMIENTO FRACCIONADOR
5.1. Nombre
5.2. Dirección exacta
5.3. País
5.4. Número de registro sanitario del establecimiento
6. FORMA FARMACÉUTICA/COSMÉTICA
7. ESPECIFICACIONES Y MÉTODOS DE CONTROL DE LOS COMPONENTES DE LA FÓRMULA.
8. METODOLOGÍA DE ELABORACIÓN DEL PRODUCTO (Describir resumidamente el proceso de fabricación).
9. ESPECIFICACIONES Y MÉTODOS DE CONTROL DEL PRODUCTO TERMINADO (Indicar y describir las especificaciones y los métodos que se utilizan en la evaluación cualitativa de los componentes de la formulación en el producto terminado).
9.1. Métodos Microbiológicos
9.2. Métodos Químicos
9.3. Métodos Físicos
9.4. Métodos Físico-químicos
10. FORMA DE PRESENTACIÓN Y CANTIDAD DE PRODUCTO
11. ESPECIFICACIÓN Y CONTROL DE ENVASES
11.1. Características del envase
11.2. Sistema de inviolabilidad
11.3. Control de calidad de envases
12. PERÍODO DE VALIDEZ (Vencimiento del lote)
13. INSTRUCCIONES DE USO
14. ESPECIES DE DESTINO
15. PRECAUCIONES GENERALES
15.1. Límite máximo y mínimo de temperatura para su correcta conservación
15.2. Describir la forma adecuada de almacenamiento
16. CAUSAS QUE PUEDEN HACER VARIAR LA CALIDAD DEL PRODUCTO
(Precipitaciones, disociaciones, frío, calor, luz, humedad, compresión en estibas o depósito).
Se adjuntan a esta solicitud los requisitos de registro establecidos en este reglamento. Toda la información que se adjunta a esta solicitud, es parte integral de la misma.
Declaramos que la información presentada es verdadera y toda alteración o información falsa, invalida esta solicitud, sin menoscabo de la responsabilidad penal que ello implica.
Nombre y firma del regente_______________
Lugar y Fecha ______
Nombre y firma del registrante ___________________
A4- FORMULARIO DE SOLICITUD DE REGISTRO SANITARIO PARA KITS DE DIAGNÓSTICO DE USO VETERINARIO
1. NOMBRE COMERCIAL DEL PRODUCTO
2. CLASIFICACIÓN (uso oficial exclusivo)
3. ESTABLECIMIENTO SOLICITANTE: PROPIETARIO/REPRESENTANTE LEGAL
3.1. Nombre
3.2. Dirección exacta
3.3. País
3.4. Número de registro sanitario del establecimiento registrante
3.5. Responsable técnico
3.5.1. Número de identificación profesional
3.5.2. Lugar o medio para recibir notificaciones
4. ESTABLECIMIENTO ELABORADOR
4.1. Nombre
4.2. Dirección exacta
4.3. País
4.4. Número de registro sanitario del establecimiento
5. ESTABLECIMIENTO FRACCIONADOR
5.1. Nombre
5.2. Dirección exacta
5.3. País
5.4. Número de registro sanitario del establecimiento
6. ESPECIFICACIONES TÉCNICAS Y COMPONENTES DEL PRODUCTO TERMINADO
6.1. Sensibilidad
6.2. Especificidad
6.3. Reproducibilidad
6.4. Límite de detección, cuando aplica
6.5. Límite de cuantificación, cuando aplica
7. FORMA DE PRESENTACIÓN (CONTENIDO NETO)
8. PERÍODO DE VALIDEZ (Vencimiento del lote)
9. INSTRUCCIONES DE USO
10. ESPECIE DE INTERÉS DIAGNÓSTICO
11. PRECAUCIONES GENERALES
11.1. Límite máximo y mínimo de temperatura para su correcta conservación.
11.2. Describir la forma adecuada de almacenamiento.
12. CAUSAS QUE PUEDEN HACER VARIAR LA CALIDAD DEL PRODUCTO (frío, calor, luz, humedad, compresión en estibas o depósito).
Se adjuntan a esta solicitud los requisitos de registro establecidos en este reglamento. Toda la información que se adjunta a esta solicitud, es parte integral de la misma.
Declaramos que la información presentada es verdadera y toda alteración o información falsa, invalida esta solicitud, sin menoscabo de la responsabilidad penal que ello implica.
Nombre y firma del regente ______________
Lugar y Fecha ______
Nombre y firma del registrante____________________
A5- FORMULARIO DE SOLICITUD DE RENOVACIÓN DE REGISTRO SANITARIO PARA MEDICAMENTOS VETERINARIOS Y PRODUCTOS AFINES
1. Nombre de la empresa solicitante
2. Número de registro sanitario de la empresa
3. Nombre del propietario o representante legal
4. Renovación (Nº de registro sanitario)
5. Dirección
6. Teléfono y fax
7. Correo electrónico
8. Lugar o medio para recibir notificaciones
9. Nombre comercial del producto
10. Fabricante
11. País de origen
12. Ciudad
13. Estado
14. Nombre del profesional (regente) que solicita el registro sanitario
15. Número de identificación profesional
16. Teléfono
17. Tel. móvil
18. Correo electrónico.
Se adjuntan a esta solicitud los requisitos de renovación de registro establecidos en este reglamento.
Toda la información que se adjunta a esta solicitud, es parte integral de la misma.
Declaramos que la información presentada es verdadera y toda alteración o información falsa, invalida esta solicitud, sin menoscabo de la responsabilidad penal que ello implica.
Nombre y firma del regente ____________
Lugar y Fecha _______
Nombre y firma del registrante __________
FIN DEL ANEXO A
ANEXO B
(NORMATIVO)
Certificado de Libre Venta (CLV)
Debe ser emitido por la autoridad competente del país de origen, constar en original y vigente, con el trámite consular correspondiente y contener la siguiente información:
Se certifica por el presente, a solicitud del gobierno de (nombre del país), que los productos de uso veterinario abajo detallados, de acuerdo con (legislación del país de origen), se fabrica(n) y comercializa(n) en (país) por (nombre del fabricante), establecido en (dirección completa), con registro sanitario Nº (número del registro sanitario del fabricante), titular del registro sanitario (nombre), establecido en (dirección completa), con registro sanitario Nº (número del registro sanitario del titular), elaborado por (establecimiento maquilador) -para en caso de maquila( titular del registro), establecido en (dirección completa), con registro sanitario Nº (número del registro sanitario del maquilador), nombre comercial, forma farmacéutica, fórmula comercial (de acuerdo con el Sistema Internacional de Unidades), indicaciones, especies de destino (especificar), Nº registro sanitario, vigencia del registro sanitario, condiciones de venta (libre venta, controlado), vigencia del documento (país, ciudad, fecha), firma y sello de la autoridad competente.
Si el CLV de países fuera del área centroamericana no contiene toda la información contemplada en este Anexo, el interesado debe presentar un documento complementario emitido por la autoridad competente del país de origen, con la información faltante.
Si el medicamento veterinario o producto afín no posee certificado de libre venta porque no se comercializa en el país de origen, el interesado debe presentar un certificado de producto destinado para la exportación, emitido por la autoridad competente que contenga al menos la información antes señalada para el CLV y la(s) razón(es) por la(s) cual(es) el mismo no se comercializa en ese país.
En el caso de que este documento no declare el origen, el interesado debe presentar adicionalmente el certificado de origen emitido por la autoridad correspondiente del país de origen.
Para los productos afines que no son objeto de registro por la autoridad competente sanitaria en el país de origen y están bajo la regulación de otra entidad, dicha autoridad sanitaria deberá emitir un documento indicando tal condición y su fundamento legal, en cuyo caso se aceptarán las certificaciones emitidas por otras entidades regulatorias del país de origen, cuando aplique.
FIN DEL ANEXO B
ANEXO C
(NORMATIVO)
INFORMACIÓN TÉCNICA DE RESPALDO PARA SOLICITAR UN REGISTRO SANITARIO DE MEDICAMENTO VETERINARIO.
A. FÁRMACOS, QUÍMICOS Y ECTOPARASITICIDAS PARA USO VETERINARIO.1. Descripción del proceso de elaboración.
2. Información del origen de las materias primas, otros ingredientes descritos en una farmacopea y materiales de acondicionamiento.
3. Pruebas de estabilidad (especificaciones para el plazo de validez, descripción de los estudios, resultados y conclusiones).
4. Estudios de eliminación de residuos o de comprobación del período de retiro y descarte, del producto a registrar, cuando aplique.
5. Para medicamentos veterinarios genéricos, adicionalmente presentar los estudios de bioequivalencia.
6. Para medicamentos con moléculas nuevas o innovadoras, excepto productos de medicina alternativa, adicionalmente presentar:
6.1. Pruebas de seguridad en la especie destino, con sus correspondientes referencias bibliográficas.
6.2. Documentación clínica: estudios de eficacia y determinación de dosis.
6.3. Estudios de efectos biológicos no deseados
6.3.1. Carcinogénicos.
6.3.2. Teratogénicos.
6.3.3. Mutagénicos
6.3.4. Resistencia a patógenos, según corresponda
6.3.5. Trastornos sanguíneos
6.3.6. Neurotoxicidad
6.3.7. Hipersensibilidad
6.3.8. Sobre la reproducción
6.3.9. Sobre la flora digestiva, según corresponda
6.3.10. Estudios de impacto ambiental realizados por el fabricante.
B. BIOLÓGICOS VETERINARIOS.
1. Definir y caracterizar las cepas (semilla maestra y líneas de producción).
2. Titulaciones o pruebas de sensibilidad.
3. Información sobre el recipiente y sistema de inviolabilidad del envase o empaque.
4. Estudios y propiedades inmunológicas cuando proceda.
5. Métodos de producción.
6. Protocolo de fabricación (preparación e incubación del organismo matriz de siembra, según corresponda); producción del organismo de siembra de trabajo, cosecha (manejo y preparación, periodo mínimo y máximo de tiempo desde la inoculación hasta la cosecha), cultivo de placas, preparación del pre-inóculo e inóculo, inactivación del antígeno, concentración del antígeno, producción de la vacuna, preparación del producto (composición del preservante y adyuvante), método y grado de concentración, información del producto, método de envasado y desecamiento, cantidad de material antígeno por dosis, pruebas (pureza, inocuidad, seguridad, potencia e identidad)
7. Información del origen de las materias primas, otros ingredientes descritos en una farmacopea y materiales de acondicionamiento.
8. Pruebas de control del producto terminado.
9. Pruebas de estabilidad y su protocolo (especificaciones para el plazo de validez, descripción de los estudios, resultados y conclusiones).
10. Para producto biológico innovador presentar, además:
10.1. Pruebas de seguridad en la especie destino, con sus correspondientes referencias bibliográficas.
10.2. Documentación clínica: estudios de eficacia y determinación de dosis.
10.3. Estudios de efectos biológicos no deseados:
10.3.1. Carcinogénicos.
10.3.2. Teratogénicos.
10.3.3. Mutagénicos
10.3.4. Trastornos sanguíneos
10.3.5. Neurotoxicidad
10.3.6. Hipersensibilidad
10.3.7. Sobre la reproducción
10.4. Estudios de impacto ambiental realizados por el fabricante.
APÉNDICE I DEL ANEXO C
CRITERIOS PARA LA COMPROBACIÓN DEL PERÍODO DE RETIRO Y/O DESCARTE DE MEDICAMENTOS VETERINARIOS
Los Estados Parte acuerdan la aplicación del literal A numeral 4 del Anexo C del RTCA Medicamentos Veterinarios, Productos Afines y sus Establecimientos. Requisitos de Registro Sanitario y Control, en su versión vigente, bajo los siguientes criterios:
1. La comprobación del período de retiro y/o descarte se debe realizar a los medicamentos veterinarios utilizados en las especies productoras de alimentos para consumo humano, una vez en la vida comercial del producto y el estudio debe ser reconocido por los demás Estados Parte.
2. El cumplimiento de la comprobación debe hacerse de acuerdo con el calendario homologado de implementación gradual, para las sustancias contenidas en el siguiente cuadro:
_________________________________________
2 Período de retiro y/o descarte: se entiende por período de retiro _ el intervalo de tiempo transcurrido entre la última administración de un medicamento veterinario a un animal, en condiciones normales de uso y el momento de sacrificio de ese animal para el consumo humano o el período durante el cual deben descartarse leche, huevos y miel, requerido para que los residuos y metabolitos del medicamento veterinario alcancen los niveles de inocuidad aceptados internacionalmente.
3. La comprobación se debe realizar por producto, no por principio activo.
4. Los Estados Parte deben revisar las listas de principios activos y grupos farmacológicos de mayor riesgo con el fin de actualizarlas, de tal forma que al final del proceso y cuando corresponda, todos los medicamentos veterinarios que se utilicen en especies productoras de alimentos cuenten con el respectivo estudio de comprobación del período de retiro y/o descarte.
5. Cuando exista evidencia de riesgo de un producto se puede realizar la reevaluación del período de retiro y/o descarte aprobado, según lo recomienda el artículo 112 literal c) de la Directriz de Codex CAC/GL 71-2009 o el que corresponda en su versión vigente.
6. Cuando haya ampliación de uso de un medicamento veterinario hacia otra especie de consumo humano, el período de retiro y/o descarte que declare, está sujeto a un estudio de comprobación para la nueva especie, excepto cuando aplique la extrapolación.
7. Cuando haya cambios en la dosificación de un medicamento veterinario registrado, el período de retiro y/o descarte está sujeto a un estudio de comprobación para la nueva dosificación.
8. Cuando haya cambios de excipientes de un medicamento veterinario registrado, el período de retiro y/o descarte que declare está sujeto a un estudio de comprobación, si el excipiente modifica las características farmacocinéticas del producto, situación que se debe respaldar con la literatura o estudios pertinentes.
9. En el caso de combinaciones de ingredientes activos a dosis fijas, la comprobación se debe hacer para aquel principio activo que tenga el período de retiro y/o descarte más prolongado, individualmente.
10. La comprobación del período de retiro y/o descarte es aplicable a los medicamentos veterinarios de nuevo registro sanitario y sujetos a renovación que no hayan aportado los estudios al momento del registro sanitario y de acuerdo con el calendario aprobado en el numeral 2 supra.
11. En los casos que un mismo fabricante elabore un producto con diferentes nombres comerciales, pero idéntico al que cuenta con el estudio de la determinación del período de retiro y/o descarte, no se requiere de un estudio individual por cada uno de los productos.
12. Si se solicita ampliar la vía de administración de un producto similar autorizada para el producto de referencia, se debe presentar un estudio de eliminación de residuos o de comprobación de periodo de retiro y/o descarte para esa nueva vía.
13. Para realizar el estudio del período de retiro y/o descarte se deben utilizar las siguientes herramientas:
a. Guía del Protocolo del Estudio.
b. Procedimiento para la implementación de la Guía del Protocolo.
FIN DEL APÉNDICE 1
APENDICE II DEL ANEXO C
PROCEDIMIENTO PARA EJECUTAR LA GUÍA DEL PROTOCOLO PARA LA COMPROBACIÓN DEL PERÍODO DE RETIRO Y/O DESCARTE DE MEDICAMENTOS VETERINARIOS
1. El interesado debe solicitar por escrito a la autoridad competente su aprobación para realizar el ensayo de comprobación del período de retiro y/o descarte y presentar los siguientes documentos:
a) Solicitud por escrito.
b) Aportar el protocolo de ensayo, para cada especie de destino o subproducto de consumo humano firmado por el médico veterinario responsable del ensayo.
c) Si el estudio es a través de un tercero, presentar la autorización o poder del fabricante para este fin y la solicitud por escrito.
2. La solicitud debe contener:
a) Nombre del producto.
b) Ingrediente activo a evaluar.
c) Especie en la que se aplica.
d) Uso terapéutico.
e) Vía de administración.
f) Dosis y duración del tratamiento.
g) Período de retiro y/o descarte a comprobar.
h) Número de registro sanitario del medicamento veterinario, cuando aplique.
i) Fecha de vencimiento del registro sanitario, cuando aplique.
j) Fabricante y país de origen.
k) Lugar donde se realiza el ensayo.
l) Persona a contactar con sus números de teléfono y correo electrónico.
m) Referencia del LMR a utilizar.
3. La autoridad competente posteriormente debe contactar al solicitante y establecer una fecha para coordinar todo lo referente al ensayo, según los plazos establecidos en cada Estado Parte.
4. El médico veterinario oficial y el médico veterinario responsable del ensayo de campo, deben realizar la inspección del establecimiento donde se va a efectuar el ensayo, verificando lo siguiente:
a) Instalaciones adecuadas de trabajo: corral, brete (prensa), manga (chutra) y báscula.
b) Identificación y registro de los animales.
c) Durante el ensayo se le deben garantizar a todos los animales las condiciones de bienestar animal, según la legislación de cada Estado Parte (manejo, alimentación y sanidad).
d) Plan de manejo sanitario adecuado.
5. El médico veterinario responsable del ensayo debe evaluar las condiciones de salud de los animales objeto del ensayo y asegurarse que no hayan sido tratados con ningún tipo de producto que pueda interferir en el estudio, según el protocolo.
6. Para la aplicación del medicamento veterinario se requiere la presencia obligatoria del médico veterinario oficial, quien debe refrendar mediante firma el formulario correspondiente.
7. Durante la ejecución del ensayo el médico veterinario oficial puede realizar visitas de verificación, en coordinación con el interesado.
8. Toma de muestras.
8.1. La toma de las muestras es realizada por el médico veterinario oficial en el establecimiento.
8.2. Se debe establecer un mecanismo que garantice la custodia e integridad de la muestra hasta su entrega al laboratorio autorizado. El mismo deberá ser consignado en el protocolo respectivo.
8.3. El interesado es el responsable de retirar las muestras y entregarlas al laboratorio autorizado respetando la integridad de las mismas.
8.4. Se debe establecer un mecanismo que garantice la custodia e integridad de la contramuestra, la cual debe almacenarse bajo custodia oficial, debiendo consignarse en el protocolo respectivo.
8.5. El interesado debe elaborar el informe final según lo establecido en el Anexo 4 del Apéndice III del Anexo C, aportando toda la documentación de respaldo en original para el expediente del registro sanitario o su renovación.
8.6. El análisis e interpretación de los resultados deben ser realizados según se indica en la Guía del Protocolo del Apéndice lll del Anexo C.
9. La autoridad competente debe comunicar por escrito al interesado los resultados según sea el caso:
a) Si el resultado es satisfactorio, se debe emitir una resolución aceptando el ensayo.
b) Si el resultado no es satisfactorio, se debe emitir una resolución comunicando el rechazo indicándole que debe realizar un estudio de eliminación de residuos completo, según la normativa internacional, para poder asignar su período de retiro y/o descarte. El interesado puede apelar esta resolución según los mecanismos internos de cada Estado Parte.
c) De no cumplir con lo solicitado en el literal b) para los productos en proceso de registro sanitario o renovación, la autoridad competente puede cancelar o denegar su registro sanitario e informar a las instancias que correspondan.
d) Los ensayos deben ser refrendados por la autoridad competente sanitaria donde se realicen.
e) Previa evaluación del caso el interesado por única vez puede readecuar su formulación, sin modificar el principio(s) activo(s) ni concentración; solo con la finalidad de corregir la cinética del medicamento veterinario y realizar un nuevo ensayo de comprobación del período de retiro y/o descarte.
FIN DEL APÉNDICE II
APÉNDICE III DEL ANEXO C
GUÍA DEL PROTOCOLO PARA LA COMPROBACIÓN DEL PERÍODO DE RETIRO Y/O DESCARTE DE MEDICAMENTOS VETERINARIOS UTILIZADOS EN ANIMALES PRODUCTORES DE ALIMENTOS PARA CONSUMO HUMANO EN LOS ESTADOSPARTE
1. Introducción
El estudio de comprobación de período de retiro y/o descarte es una prueba en un único punto de comprobación en el tejido marcador teniendo validez técnica, pero circunscrita a la comparación de los resultados del período de retiro y/o descarte con el medicamento veterinario de referencia o bioequivalente, en el cual se utilizan menos animales y el resultado sólo indica si la comprobación es aceptada. Esta prueba sólo establece, si los niveles de residuos aún después de aplicar una tolerancia del 95% (UTL 95%), presenta valores iguales o inferiores del Límite Máximo de Residuos (LMR) permitido en el punto (en el tiempo) que se ha seleccionado para la prueba. No calcula el período de retiro y/o descarte, pero sí confirma un período específico de retiro y/o descarte, comparado con el medicamento veterinario de referencia o bioequivalente.
2. Antecedentes
Según la legislación de cada Estado Parte donde se realice el estudio.
3. Criterios para determinar la necesidad de un estudio de residuos o de un estudio de comprobación de período de retiro y/o descarte
Los criterios para determinar cuáles medicamentos veterinarios deben ser sometidos a un estudio de comprobación de período de retiro y/o descarte, depende del tipo de producto farmacológico veterinario similar de que se trate, del (los) principio(s) activo(s) presentes e indicaciones de uso, donde se deben tomar en cuenta las especificaciones descritas a continuación.
Las autoridades competentes deben evaluar la pertinencia y validación de las metodologías analíticas utilizadas.
Los estudios de residuos o de comprobación del período de retiro y/o descarte, deben realizarse para medicamentos veterinarios utilizados en animales cuyos productos y subproductos sean destinados al consumo humano.
3.1 Medicamentos veterinarios que NO REQUIEREN realización de estudios de comprobación de período de retiro y/o descarte en un único punto:
Se excluyen de este tipo de estudios los siguientes grupos:
a) Medicamentos veterinarios genéricos o similares con formulaciones idénticas, o con la misma cantidad de ingrediente activo del medicamento de referencia; en los que se ha demostrado bioequivalencia.
b) Medicamentos veterinarios que demuestren que la biodisponibilidad de su(s) principio(s) activo(s) en la(s) especie(s) de destino sea menor o igual a la del producto de referencia y cuyo período de retiro y/o descarte sea de 0 a 2 días.
3.2 Medicamentos veterinarios que REQUIEREN como mínimo un estudio de comprobación de período de retiro y/o descarte (en un único punto):
a) Medicamentos veterinarios genéricos/similares donde en las pruebas de bioequivalencia en la(s) especie(s) de destino, la biodisponibilidad del principio activo se mantiene superior a la del producto de referencia, durante todo el período de retiro y/o descarte recomendado para el producto de referencia.
b) Medicamentos veterinarios similares que no hayan demostrado bioequivalencia con el producto de referencia.
c) Medicamentos veterinarios en los cuales se ha modificado alguno de los excipientes y que se tiene seguridad que altera la biodisponibilidad del ingrediente activo en la(s) especie(s) de destino en los tejidos marcadores.
4. Interpretación de los resultados del estudio de comprobación de período de retiro y/o descarte:
a) Si el estudio demuestra que el límite superior de tolerancia unilateral del 95% (UTL) presenta valores de residuos iguales o inferiores al correspondiente LMR, la comprobación es aceptada. La prueba estadística a utilizar es:
UTL = media ± kSDm.
UTL =Límite Superior de Tolerancia a la media en el punto único.
SDm = desviación estándar (típica) de la muestra.
k = Constante que se toma de la tabla del Límite de Factor de Tolerancia unilateral del 95% (percentil con una confianza del 95%), para una distribución normal
Shapiro-Wilk: herramienta estadística que se utiliza para determinar la normalidad de los datos obtenidos del estudio de comprobación del período de retiro y/o descarte.
Cuando se realiza la evaluación de los datos y éstos no presentan una distribución normal según la fórmula Shapiro-Wilk, la metodología antes señalada para la comprobación del período de retiro y/o descarte no es aplicable, por tanto, se debe usar un método alternativo que consiste en incrementar el período de retiro y/o descarte del producto de referencia en un rango entre 10-30% dependiendo del riesgo del producto, según EMA/CVMP/036/95, pág. 11 y 12.
b) Si el estudio demuestra lo contrario, es decir, que los residuos están por encima del LMR entonces la prueba no se acepta y no se puede utilizar el período establecido por el producto de referencia por lo que debe realizarse un estudio de depleción de residuos.
c) Métodos de análisis y evaluación: se deben incluir los métodos de análisis y evaluación del principio activo. La metodología debe ser de referencia (CODEX-FDA-EMA) o estar validada científicamente (se debe aportar el método analítico completo con su correspondiente validación).
d) El punto de medición a considerar es el último tiempo (día, horas, entre otros) del período de retiro y/o descarte del medicamento veterinario de referencia o bioequivalente.
e) Cronograma de actividades y ejecución: programar todas las actividades que deben ser desarrolladas para cumplir con el objetivo del estudio.
f) La cantidad de animales o productos de origen animal debe ser según se describe en la tabla siguiente:
tabla 1*
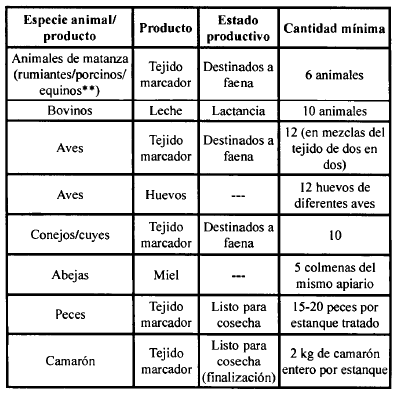
* Según referencia Guía Técnica del Comité de las Américas de Medicamentos Veterinarios (CAMEVET), para la Conducción de Estudios de Metabolismo y Cinética de Residuos de Agentes Farmacológicos de Uso Veterinario en Animales Productores de Alimentos. Estudios de eliminación de residuos para establecer períodos de retiro y/o descarte del medicamento veterinario.
**En aquellos Estados Parte donde los equinos sean destinados al consumo humano o animal.
5. Informe general:
Fecha de inicio del ensayo: ------------------------------------
Fecha de finalización del ensayo: _________________________________
Producto:
Nombre comercial del producto: __________________________________
Principio activo y concentración: __________________________________
Fabricante: ___________________________________________
País de origen: _______________________________________________
Importador y/o distribuidor: ______________________________________
Fecha y fumigación del producto: _________________________________
Fecha de expiración del producto: _________________________________
Número de lote: ________________________________________________
Presentación: __________________________________________________
Especie destino o subproductos destinados al consumo humano: _________
Vía de administración o aplicación: __________________________________
Dosis y duración del tratamiento: ____________________________________
Período de retiro y/o descarte: ______________________________________
Finca o explotación:
Nombre de la explotación: _________________________________________
País: __________________________________________________________
Propietario: _____________________________________________________
Dirección del propietario: __________________________________________
Teléfono: ________________ Correo electrónico: ______________________
Dirección georreferenciada de la finca: _______________________________
Animales tratados:
Tiempo de resguardo del producto: ________________________ (días, horas, entre otros).
Tiempo que duró el tratamiento: _________________________ (días, horas, entre otros).
Fecha de aplicación del producto: _____________________________________________
No. de animales tratados: ___________________ Vía de aplicación: __________________
Fecha de envío al sacrificio: __________________________________________________
Toma de muestra en matadero:
Fecha de recibido de los animales en el matadero: ________________________________
Fecha de sacrificio: ________________Hora: _______________________
Fecha de toma de las muestras: __________________________________
No. de muestras recolectadas: ___________________________________
Tejido marcador donde se toma la muestra: _________________________
Persona responsable de tomar las muestras: ________________________
Nombre del médico veterinario oficial responsable: ___________________
Responsable del retiro y/o descarte de las muestras: _________________
Medio de conservación de la muestra: _____________________________
Fecha de retiro y/o descarte de las muestras: _______________________
Laboratorio;
Nombre del laboratorio autorizado: ________________________________
Institución a que pertenece el laboratorio: ___________________________
Fecha y hora de recibido de las muestras en el laboratorio: _____________
Temperatura de recibido de la muestra en el laboratorio. _______________
Análisis e interpretación de los resultados:
Entidad que realizó el análisis: __________________________________
Nombre del responsable del ensayo: _____________________________
Firma: __________
________________________________________________________________
Visto bueno de la autoridad competente:
Nombre _____________ Firma: ______________
6. Guía para la realización del estudio:
6.1 Requisitos para las pruebas de comprobación de período de retiro y/o descarte:
Aspectos generales
Para la realización de las pruebas de comprobación de período de retiro y/o descarte se deben cumplir con las siguientes directrices:
a) El protocolo para la realización del ensayo debe ser aprobado por la autoridad competente.
b) Utilizar metodologías que permitan predecir el comportamiento del medicamento veterinario en la especie de destino.
c) Los laboratorios o instituciones, así como los profesionales responsables por la ejecución de las pruebas analíticas deben estar previamente autorizados por la autoridad competente, para la realización de esa prueba y cumplir con los principios éticos en relación con la confidencialidad y transparencia.
d) Las instituciones, laboratorios o empresas responsables por la ejecución de la prueba analítica deben tener experiencia comprobada para la realización de la misma, así como las instalaciones y equipos necesarios de acuerdo con la metodología a ser utilizada.
e) Se deben realizar tantos estudios de comprobación de residuos, como especies destinadas a la alimentación humana tenga el medicamento veterinario a analizar y si es pertinente, incluir la extrapolación de especies utilizando guías de extrapolación internacionalmente aceptadas.
f) La autoridad competente del país donde se realice el ensayo es la encargada de evaluar y aprobar los protocolos de los estudios de comprobación de período de retiro y/o descarte y debe participar de la supervisión de la prueba, al menos en las fechas de aplicación del producto y en el muestreo de los tejidos o productos.
Objetivo
Establecer los criterios o principios para la realización de estudios de comprobación del período de retiro y/o descarte de los medicamentos veterinarios que lo requieran en los Estados Parte.
6.2 Metodología del estudio
6.2.1 Normas de referencia
a) Guía Técnica del Comité de las Américas de Medicamentos Veterinarios (CAMEVET), para la Conducción de Estudios de Metabolismo y Cinética de Residuos de Agentes Farmacológicos de Uso Veterinario en Animales Productores de Alimentos. Estudios de eliminación de residuos para establecer períodos de retiro y/o descarte del medicamento veterinario.
b) Guía VICH GL 2, GL48, GL 49.
6.2.2 Medicamento veterinario objeto de estudio
Descripción del medicamento veterinario en estudio.
6.2.3 Especie(s) o producto(s) en que se realiza el estudio
Según la especie(s) o producto(s) del ensayo.
6.2.4 Condición sanitaria de los animales y explotaciones
6.2.4.1 Características y condiciones sanitarias de los animales objeto del estudio
a) Animales representativos de la especie en la que se está indicando el producto.
b) Buen estado de salud (examen clínico).
c) Animales sin signos compatibles con enfermedades.
d) Buena condición corporal.
e) Animales de matanza o en las distintas etapas de producción según se trate el producto que se vaya a comprobar su período de retiro y/o descarte.
f) Los animales sometidos a la evaluación no deben haber recibido tratamiento con ningún medicamento veterinario que interfiera con los resultados del estudio.
6.2.4.2 Características de la explotación o la finca
a) Instalaciones adecuadas de trabajo: corral, brete (prensa), manga (chutra) y báscula.
b) Identificación y registro de los animales. Los animales deben estar identificados de forma segura.
c) La finca debe tener un sistema de manejo general que permita una adecuada alimentación de los animales con base en sus requerimientos nutricionales.
6.2.4.3 Número de animales necesarios para el estudio
El número de animales según la Tabla 1.
6.2.4.4 Medicamento veterinario a evaluar
Los medicamentos veterinarios a utilizar deben ser suministrados por el interesado, garantizando que han sido almacenados de acuerdo con lo que establece la etiqueta del producto.
6.2.4.5 Aplicación del medicamento veterinario
a) Los animales deben ser pesados de forma que la dosis del medicamento veterinario administrado sea precisa, según la dosis máxima recomendada.
b) La aplicación del producto debe ser por un médico veterinario responsable del ensayo, por la vía utilizada para establecer el periodo de retiro del producto de referencia, bajo la supervisión directa de un médico veterinario oficial designado.
c) Se debe aplicar la dosis máxima declarada por el fabricante en el etiquetado y el tratamiento debe ser sobre la máxima duración prevista.
d) Los envases utilizados de los productos aplicados deben quedar bajo la custodia de la autoridad competente, hasta la finalización del estudio.
6.2.4.6 Duración del ensayo y recopilación de información.
a) El ensayo tiene una duración correspondiente al período de retiro y/o descarte del medicamento veterinario de referencia.
b) Durante el ensayo se le deben garantizar a todos los animales las condiciones de bienestar animal (manejo, alimentación y sanidad).
c) Durante el ensayo no se debe aplicar ningún medicamento veterinario que pueda afectar los resultados de la prueba.
d) Cualquier animal que presente cualquier signo clínico durante el ensayo, debe ser excluido del mismo.
e) Durante la realización del ensayo, la información debe ser recabada por los técnicos del laboratorio solicitante del estudio y refrendada por el médico veterinario oficial designado por la autoridad competente.
f) La realización del ensayo se debe efectuar de lunes a viernes, en horas laborales del médico veterinario oficial designado por la autoridad competente.
6.2.4.7 Sacrificio y toma muestra
a) Los animales deben ser sacrificados en un solo día, en plantas de proceso y los subproductos en establecimientos de producción primaria, donde las muestras y contramuestras deben ser recolectadas bajo la supervisión del médico veterinario oficial.
b) La identificación de las muestras y contramuestras, deben mantener la trazabilidad respectiva.
c) Tipo de muestras: tejido marcador o subproducto, según el medicamento veterinario a evaluar de acuerdo con el Anexo 3.
d) Peso mínimo de la muestra y contramuestra, requerido según la especie y el ensayo.
6.2.5 Resultados e informe final del estudio
a) Al momento de emitir los resultados, el laboratorio debe remitir el original de los mismos al interesado y copia a la autoridad competente.
b) El interesado debe elaborar el informe final según lo establecido en el Anexo 4, aportando toda la documentación de respaldo en original, para el expediente de registro o renovación del producto.
6.2.6 Costos del ensayo
Los costos asociados al ensayo deben ser asumidos por el interesado.
ANEXO 1
LISTADO DE ANIMALES A TRATAR
PARA LA PRUEBA DE COMPROBACIÓN DE RESIDUOS
Fecha del pesaje: ________
Especie: ________________
Raza: ___________________
ANEXO 4
APARTADOS A DESARROLLAR EN EL INFORME FINAL DE LA COMPROBACIÓN DEL PERÍODO DE RETIRO Y/O DESCARTE DE MEDICAMENTOS VETERINARIOS UTILIZADOS EN ANIMALES PRODUCTORES DE ALIMENTOS PARA CONSUMO HUMANO EN LOS ESTADOS PARTE
El informe final debe contener los siguientes apartados:
1. Título.
2. Autor(es) del estudio
3. Resumen en español e inglés u otro idioma
4. Introducción
5. Materiales y métodos
6. Resultados y discusión
7. Conclusiones
8. Referencias bibliográficas
FIN DEL APÉNDICE III
FIN DEL ANEXO C
--------------------------------
ANEXO D
(NORMATIVO)
ELEMENTOS A APORTAR CUANDO APLIQUE EL CONTRATO DE FABRICACIÓN O MAQUILA
1. Identificación de las partes contratantes facultadas para suscribir un contrato, conforme a la legislación de cada Estado Parte.
2. Las obligaciones y responsabilidades de cada una de las partes, en relación con la fabricación, manejo, almacenamiento, liberación, etiquetado, re-envase y/o re-empaque, del producto, cuando aplique.
3. Las responsabilidades de cada una de las partes, en relación con la calidad, seguridad y eficacia del producto.
4. Manifestación expresa de la aceptación de los acuerdos pactados por las partes respecto a los puntos señalados en el presente anexo.
5. Lista de cada uno de los productos, la cual debe anexarse y puede modificarse cuando sea necesario.
FIN DEL ANEXO D
--------------------------------
ANEXO E
(NORMATIVO)
 TIPOLOGÍA Y REQUISITOS PARA LAS MODIFICACIONES AL REGISTRO SANITARIO (ANOTACIONES MARGINALES)
--------------------------------
ANEXO F
(NORMATIVO)
REQUISITOS PARA SOLICITAR LA EXENCIÓN DE REGISTRO SANITARIO DE MEDICAMENTOS VETERINARIOS Y PRODUCTOS AFINES CON FINES DE INVESTIGACIÓN EN LOS ESTADOS PARTE
TIPOLOGÍA Y REQUISITOS PARA LAS MODIFICACIONES AL REGISTRO SANITARIO (ANOTACIONES MARGINALES)
--------------------------------
ANEXO F
(NORMATIVO)
REQUISITOS PARA SOLICITAR LA EXENCIÓN DE REGISTRO SANITARIO DE MEDICAMENTOS VETERINARIOS Y PRODUCTOS AFINES CON FINES DE INVESTIGACIÓN EN LOS ESTADOS PARTE
El interesado debe aportar los siguientes documentos a la autoridad competente:
1. Solicitud escrita del interesado con la debida justificación técnica que motiva la realización del ensayo en el Estado Parte respectivo.
2. Adjuntar el protocolo de ensayo según lo establecido en el Anexo L del presente reglamento y la documentación técnica de respaldo.
3. Cumplir con las disposiciones que establece cada Estado Parte en relación con el uso de animales de experimentación para fines de investigación.
4. En caso de que el producto deba importarse, se debe indicar en el protocolo de ensayo la cantidad de producto a importar y cumplir con los trámites de importación dispuestos en cada Estado Parte.
Una vez aprobada la solicitud y el protocolo de ensayo, el interesado puede iniciar con el respectivo ensayo según lo establecido en el mismo.
Al finalizar el ensayo, el interesado debe presentar un informe final a la autoridad competente con los resultados obtenidos y las respectivas conclusiones.
FIN DEL ANEXO F
--------------------------------
ANEXO G
(NORMATIVO)
LISTADO DE MEDICAMENTOS VETERINARIOS Y PRODUCTOS AFINES DE CONFORMIDAD CON SU NIVEL DE RIESGO
El listado de los productos incluidos en cada uno de los grupos indicados en el reglamento RTCA Medicamentos Veterinarios, Productos Afines y sus Establecimientos. Requisitos de Registro Sanitario y Control, en su versión vigente, será definido y publicado por la autoridad competente de los Estados Parte. La misma sirve para la implementación de la fiscalización y farmacovigilancia de acuerdo con la capacidad y los recursos de cada Estado Parte.
FIN DEL ANEXO G
--------------------------------
ANEXO H
(NORMATIVO)
CODIFICACIÓN DE MEDICAMENTOS VETERINARIOS Y PRODUCTOS AFINES.
La codificación de medicamentos veterinarios y productos afines se detalla de la siguiente manera:
1. Para medicamentos veterinarios anteponer al número correlativo las siglas MV-.
2. Para productos afines anteponer al número correlativo las siglas AV-.
3. Número correlativo.
FIN DEL ANEXO H
--------------------------------
ANEXO I
(NORMATIVO)
CÓDIGO DE BUENAS PRÁCTICAS DE MANUFACTURA DE MEDICAMENTOS VETERINARIOS Y PRODUCTOS AFINES, GUÍA DE VERIFICACIÓN Y PLAZO DE IMPLEMENTACIÓN
Guatemala, El Salvador, Honduras, Nicaragua, Costa Rica y Panamá acuerdan utilizar como referencia para su cumplimiento, el Código de Buenas Prácticas de Manufactura de Medicamentos Veterinarios del Comité para las Américas de Medicamentos Veterinarios de OIE, así como su Guía de Verificación, ambos en su versión vigente. Para los productos biológicos veterinarios de los Estados Parte, acuerdan utilizar como referencia para su cumplimiento, normas de Buenas Prácticas de Manufactura para productos biológicos expedidas por una entidad reconocida u organismo internacional reconocido, hasta que se desarrolle la normativa específica en la materia. Para los productos afines, se aplicará la normativa de Buenas Prácticas de Manufactura que defina cada Estado Parte.
FIN DEL ANEXO I
--------------------------------
ANEXO J
(NORMATIVO)
LISTADO DE MEDICAMENTOS VETERINARIOS Y PRODUCTOS AFINES SUJETOS A REGISTRO SIMPLIFICADO
Los Estados Parte acuerdan adoptar el siguiente listado y requisitos para el registro de medicamentos veterinarios y productos afines:
a. Absorbentes y adsorbentes.
b. Antisépticos.
c. Descornadores químicos.
d. Desinfectantes.
e. Desodorantes y odorizantes.
f. Kits de diagnóstico de enfermedades.
g. Lubricantes obstétricos.
h. Marcadores quemantes.
i. Productos de higiene y belleza.
j. Repelentes no plaguicidas.
k. Rubefacientes.
l- Ungüentos y cremas tópicas que no contengan principios activos de medicamentos veterinarios de venta exclusiva en farmacias veterinarias. El listado, puede ser actualizado por los Estados Parte cuando sea técnicamente necesario.
FIN DEL ANEXO J
--------------------------------
ANEXO K
(NORMATIVO)
LISTADO DE PRODUCTOS DE USO VETERINARIOS NO SUJETOS A REGISTRO
Los productos de uso veterinario incluidos en la siguiente lista no son sujetos de registro sanitario en los Estados Parte:
1) Accesorios para mascotas
2) Arenas higiénicas para mascotas
3) Collar de adiestramiento para el control de la conducta animal
4) Crayón o marcador para ganado
5) Diluyente de semen sin ingrediente activo
6) Equipo médico y sus reactivos
7) Estabilizadores o indicadores de agua que se utilizan para aplicación de vacunas
8) Insumos médicos
9) Equipos para peluquería
10) Gel para palpación sin ingrediente activo
11) Instrumental quirúrgico o de uso en laboratorio
12) Jaulas transportadoras de mascotas
13) Kits rápidos de química sanguínea
14) Nitrógeno líquido para la conservación de biológicos, semen o embriones
15) Parches para detección de celo
16) Productos para conservar la calidad de agua en especies acuícolas ornamentales
17) Productos para manejo y trasporte de material reproductivo
18) Reactivos químicos para uso en laboratorio (no incluye los utilizados para el diagnóstico de enfermedades)
19) Selladores de pezones sin ingrediente activo
20) Tinta para tatuaje
21) Toallitas húmedas de uso veterinario sin ingrediente activo.
El listado puede ser actualizado por los Estados Parte cuando sea técnicamente necesario.
La exención de registro sanitario no implica la eliminación de los controles sanitarios oficiales realizados por cada Estado Parte.
FIN DEL ANEXO K
--------------------------------
ANEXO L
(NORMATIVO)
INFORMACIÓN BÁSICA PARA ELABORAR UN PROTOCOLO DE ENSAYO DE MEDICAMENTOS VETERINARIOS Y PRODUCTOS AFINES
La autoridad competente de los Estados Parte, debe evaluar los protocolos de ensayo clínico presentados por los fabricantes, para medicamentos veterinarios y productos afines, que contengan la siguiente información básica:
1. Introducción: descripción de la importancia del ensayo, relatando la incidencia en el país de la(s) enfermedad (es) que controla o previene el producto a evaluar y el impacto económico que esto representa (bases teóricas del ensayo).
2. Características del producto: Nombre comercial, principio activo, indicaciones, forma farmacéutica, vía de administración, dosis, especies destino, mecanismo de acción, propiedades del producto.
3. Objetivos del ensayo: - General: - Específico(s):
4. Hipótesis (si las hay): Descripción de las presunciones del producto respecto a su eficacia o inocuidad.
5. Materiales y métodos: Establecimientos donde se va a efectuar el ensayo, especies, categoría, número de animales, criterios de selección, métodos de muestreo, equipo e instrumentos a utilizar, evaluación y control inicial de los animales, fecha de inicio y finalización del ensayo, método de aplicación, control y seguimiento. Personal responsable a cargo de las actividades. Laboratorios de análisis y fichas técnicas de registro. Métodos estadísticos utilizados para la evaluación, análisis y presentación de resultados.
6. Análisis económico. Desglose de costos del ensayo.
7. Cronograma de actividades: Definidas en etapas a desarrollar, con fechas previstas para su ejecución.
8. Bibliografía.
9. Nombre, firma y calidades del investigador.
FIN DEL ANEXO L
--------------------------------
ANEXO M
(NORMATIVO)
PROCEDIMIENTO DE RECONOCIMIENTO DE REGISTRO SANITARIO DE MEDICAMENTOS VETERINARIOS Y PRODUCTOS AFINES EN LA REGIÓN CENTROAMERICANA
2. Ámbito de aplicación
Este procedimiento aplica únicamente a los productos originarios de los Estados Parte.
2. Requisitos para el reconocimiento del registro sanitario.
2.1. Solicitud de reconocimiento del registro ante la autoridad competente, firmada y sellada por el regente veterinario y el registrante.
2.2. Poder notariado del titular otorgado a favor del registrante autorizándolo a realizar estas actividades según lo establecido por la autoridad competente de cada Estado Parte.
NOTA 1. Si el poder de representación otorgado previamente se encuentra vigente y faculta al apoderado para registrar, renovar, modificar, cancelar el registro del producto y cualquier otra actividad relacionada con este, no será necesario presentar nuevamente el poder al momento de la solicitud de cualquiera de las anteriores, caso contrario deberá aportarlo.
2.3. Copia del formulario de solicitud de registro (Anexo A A1, A2, A3 o A4, según corresponda), presentada por el interesado a la autoridad competente del país donde se registró inicialmente, con la firma y sellos de las autoridades responsables.
2.4. Certificado de libre venta, en original, con el trámite consular correspondiente, según Anexo B.
2.5. Haber cumplido con los requisitos solicitados en el presente reglamento para el registro sanitario común incluyendo medicamentos veterinarios en combinaciones a dosis fijas o para el registro sanitario simplificado, según corresponda.
2.6. Etiqueta, estuche e inserto si es el caso, en original, aprobada según los requisitos de este reglamento, con la firma y sellos de la autoridad competente y su fecha de aprobación.
2.7. Copia de certificado y metodología de análisis para el producto terminado, presentada por el interesado a la autoridad competente del país donde se registró inicialmente, con la firma y sellos de las autoridades responsables.
2.8. Cuando en el proceso de fabricación se involucren dos o más laboratorios, debe especificarse el proceso en que interviene cada uno y aportar la correspondiente certificación de cumplimiento de las buenas prácticas de manufactura de cada laboratorio de acuerdo con lo establecido en el presente reglamento.
2.9. Pago del servicio, cuando corresponda.
2.10. Con fundamentación técnica, la autoridad competente de cada Estado Parte, puede solicitar al fabricante pruebas de eficacia y seguridad locales.
3. Requisitos de renovación del reconocimiento al registro sanitario
3.1. Poder notariado del titular otorgado a favor del registrante autorizándolo a realizar estas actividades según lo establecido por la autoridad competente de cada Estado Parte.
NOTA 1. Si el poder de representación otorgado previamente se encuentra vigente y faculta al apoderado para registrar, renovar, modificar, cancelar el registro del producto y cualquier otra actividad relacionada con este, no será necesario presentar nuevamente el poder al momento de la solicitud de cualquiera de las anteriores, caso contrario deberá aportarlo.
3.2. Solicitud para la renovación del reconocimiento de registro firmada y sellada por el regente veterinario y el titular o su representante legal, ante la autoridad competente del Estado Parte.
3.3. Certificado de libre venta, en original, con el trámite consular correspondiente según Anexo B.
3.4. Documento legal ante notario emitido por el titular del registro, el cual debe indicar que el registro sanitario del producto no ha sufrido ningún cambio legal, técnico ni científico desde la última solicitud de modificación aprobada por la autoridad competente.
3.5. Comprobante de pago, si corresponde.
4. Requisitos de las modificaciones al reconocimiento al registro sanitario
4.1. Poder notariado del titular otorgado a favor del registrante autorizándolo a realizar estas actividades según lo establecido por la autoridad competente de cada Estado Parte.
NOTA 1. Si el poder de representación otorgado previamente se encuentra vigente y faculta al apoderado para registrar, renovar, modificar, cancelar el registro del producto y cualquier otra actividad relacionada con este, no será necesario presentar nuevamente el poder al momento de la solicitud de cualquiera de las anteriores, caso contrario deberá aportarlo.
4.2. Pueden realizarse únicamente en el país de origen, pudiendo ser reconocidas en los Estados Parte a través de certificación emitida por la autoridad competente.
4.3. Se consideran anotaciones marginales las descritas en el Anexo E del RTCA Medicamentos Veterinarios, Productos Afines y sus Establecimientos. Requisitos de Registro Sanitario y Control, en su versión vigente.
4.4. Requisitos:
4.4.1. Solicitud para el reconocimiento de la modificación del registro, firmada y sellada por el regente veterinario y el titular del registro o su representante legal, ante la autoridad competente del Estado Parte.
4.4.2. Copia legalizada del documento original que respaldó el cambio, firmada y sellada por la autoridad competente, así como el de su debida aprobación y adjuntar el material de empaque aprobado, si procede.
4.4.3. Comprobante de pago, si corresponde.
5. Procedimiento para el reconocimiento de registro sanitario, su renovación y sus modificaciones o anotaciones marginales
5.1. Presentar los requisitos establecidos ante la autoridad competente.
5.2. La autoridad competente verifica los requisitos presentados.
5.3. La autoridad competente resuelve la solicitud mediante resolución, debiendo notificarla al interesado en el medio señalado. En caso de denegarse, indicar las razones correspondientes, pudiendo el interesado impugnar de acuerdo con lo establecido en la legislación interna de cada Estado Parte.
5.4. En caso de aprobación del reconocimiento de registro sanitario, la autoridad competente le asigna un número de registro.
5.5. El proceso concluye cuando el interesado aporta la etiqueta final con el número de registro sanitario asignado.
5.6. Una vez inscrito el producto por reconocimiento, la autoridad competente extiende el certificado de registro sanitario.
5.7. En caso de que se apruebe el reconocimiento, se debe mantener la vigencia del registro sanitario otorgada por el primer Estado Parte donde se registró el producto.
6. Causas de denegación del reconocimiento, su renovación y modificaciones o anotaciones marginales
6.1. El reconocimiento de registro de un medicamento veterinario o producto afín no debe otorgarse en los siguientes casos:
6.2. Fármacos, químicos o biológicos y sus mezclas prohibidos en el país de destino.
6.3. Su uso y manipulación represente riesgo comprobado para la salud pública, salud animal y el ambiente.
6.4. Se detecte alguna irregularidad, fraude o falsedad en la información aportada para el reconocimiento del registro.
6.5 Sustancias con indicaciones de uso no aceptadas en el país de destino del reconocimiento.
6.6 No cumplir con los requisitos establecidos en el presente reglamento.
7. Vigencia del reconocimiento de registro
El registro reconocido mantiene la vigencia que tenga el registro en el país de origen.
8. Otras consideraciones.
8.1. Si la autoridad competente de un Estado Parte cancela el registro de un producto, debe comunicarlo de manera oficial e inmediata a los demás Estados Parte.
8.2. El solicitante del reconocimiento de registro debe estar inscrito ante la autoridad competente de acuerdo con lo establecido en el reglamento RTCA Medicamentos Veterinarios, Productos Afines y sus Establecimientos. Requisitos de Registro Sanitario y Control, en su versión vigente.
8.3. No se permite comercializar un producto sin haber notificado o aprobado, según corresponda, las modificaciones realizadas al registro original.
8.4. Las solicitudes y aprobaciones para el reconocimiento se realizan por producto.
8.5. La autoridad competente puede solicitar aclaraciones sobre el expediente presentado cuando lo considere necesario.
NOTA: En DEFINICIONES el punto 3, se encuentra repetido el numeral 3.19, 3.19 Forma cosmética y 3.19 Fórmula cuali-cuantitativa, error en la publicación de La Gaceta, Diario Oficial.
FIN DEL REGLAMENTO TÉCNICO.-