Enlace a Legislación Relacionada
(APROBAR EL REGLAMENTO TÉCNICO CENTROAMERICANO RTCA 11.03.42:07 PRODUCTOS FARMACÉUTICOS. MEDICAMENTOS DE USO HUMANO. BUENAS PRÁCTICAS DE MANUFACTURA PARA LA INDUSTRIA FARMACÉUTICA Y SU GUÍA DE VERIFICACIÓN, EN LA FORMA QUE APARECE EN EL ANEXO DE LA PRESENTE RESOLUCIÓN)
RESOLUCIÓN N°. 339-2014 (COMIECO-LXVII), aprobada el 25 de abril de 2014
Publicada en La Gaceta, Diario Oficial N°. 209 del 04 de noviembre de 2014
RESOLUCIÓN No. 339-2014 (COMIECO-LXVII)
EL CONSEJO DE MINISTROS DE INTEGRACIÓN ECONÓMICA
CONSIDERANDO:
Que de conformidad con los artículos 38, 39 Y 55 del Protocolo al Tratado General de Integración Económica Centroamericana Protocolo de Guatemala, modificado por la Enmienda del 27 de febrero de 2002, el Consejo de Ministros de Integración Económica tiene bajo su competencia los asuntos de la Integración Económica Centroamericana y, como tal, le corresponde aprobar los actos administrativos del Subsistema Económico;
Que de acuerdo con el artículo 15 de ese mismo instrumento jurídico regional, los Estados Parte tienen el compromiso de constituir una Unión Aduanera entre sus territorios, la que se alcanzará de manera gradual y progresiva, sobre la base de programas que se establezcan al efecto, aprobados por consenso;
Que, en el marco del proceso de conformación de la Unión Aduanera, los Estados Parte han alcanzado importantes acuerdos en materia de Buenas Prácticas de Manufacturas para la Industria de Medicamentos de Uso Humano, que requieren la aprobación del Consejo;
Que los Estados Parte, en su calidad de Miembros de la Organización Mundial del Comercio (OMC), notificaron al Comité de Obstáculos Técnicos al Comercio, de conformidad con lo establecido en el Acuerdo sobre Obstáculos Técnicos al Comercio, el Proyecto de Reglamento Técnico Centroamericano RTCA 11.03.42:07 Productos Farmacéuticos. Medicamentos de Uso Humano. Buenas Prácticas de Manufactura para la Industria Farmacéutica;
Que los Estados Parte, concedieron un plazo prudencial a los Estados Miembros de la OMC para hacer observaciones al proyecto de Reglamento notificado tal y como lo exige el numeral 4), párrafo 9 del artículo 2 del Acuerdo sobre Obstáculos Técnicos al Comercio, observaciones que fueron debidamente analizadas y atendidas en lo pertinente;
Que, de conformidad con el Acuerdo sobre Obstáculos Técnicos al Comercio, los Miembros preverán un plazo prudencial entre la aprobación de los reglamentos técnicos y su entrada en vigor, con el fin de dar tiempo a los productores para adaptar sus productos o sus métodos de producción a lo establecido en los reglamentos;
Que de conformidad con el párrafo 3 del artículo 55 del Protocolo de Guatemala, se recabó la opinión del Comité Consultivo de Integración Económica,
POR TANTO:
Con fundamento en lo dispuesto en los artículos 1, 3, 5, 7, 15,26, 30, 36, 37, 38, 39, 46, 52 Y 55 del Protocolo al Tratado General de Integración Económica Centroamericana -Protocolo de Guatemala,
RESUELVE:
1. Aprobar el Reglamento Técnico Centroamericano RTCA 11.03.42:07 PRODUCTOS FARMACÉUTICOS MEDICAMENTOS DE USO HUMANO BUENAS PRÁCTICAS DE MANUFACTURA PARA LA INDUSTRIA FARMACÉUTICA Y SU GUÍA DE VERIFICACIÓN en la forma que aparece en el Anexo de la presente Resolución y que forma parte integrante de la misma.
2. A partir de la vigencia del RTCA 11.03.42:07 Productos Farmacéuticos Medicamentos de Uso Humano. Buenas Prácticas de Manufactura para la Industria Farmacéutica y su Guía de Verificación, se establece un período transitorio de 2 años, durante el cual se deberá cumplir el 80% de cada uno de los puntos críticos, mayores y menores, de la Guía de Verificación anexa al Reglamento. A partir del tercer año de vigencia, deberá cumplirse con al menos el 90% de cada uno de los puntos críticos, mayores y menores de dicha Guía. En el caso de Panamá durante este periodo transitorio de 2 años solicitará el 100% de los puntos críticos y el 80% en mayores y menores.
Para el caso de El Salvador, Guatemala, Nicaragua y Panamá se exigirá el cumplimiento del 100% de los puntos críticos, así como los mayores y menores incluidos en la guía en un plazo de 3 años, a partir de la vigencia del Reglamento.
Sin perjuicio de lo dispuesto en los párrafos anteriores, los Estados Parte podrán adelantarse en el cumplimiento de los plazos establecidos en esta Resolución.
No obstante, lo anterior, los plazos transitorios no aplicarán en el caso de Costa Rica, país en el que se deberá cumplir el 100% de los puntos críticos, así como los mayores y menores de la Guía de Verificación, a partir de la entrada en vigor de este Reglamento.
En el caso de Honduras, para la evaluación del cumplimiento del Reglamento se aplicará la Guía de Verificación en base a Gestión de Riesgo, al mismo tiempo se compromete a publicar en la página web, los certificados emitidos y enviar los informes de las inspecciones de Buenas Prácticas de Manufactura a solicitud de las Autoridades Reguladoras Nacionales de los Estados Partes.
Para efectos del registro y procedimiento de reconocimiento de registro, Honduras aceptará los certificados de Buenas Prácticas de Manufactura emitidos por las autoridades competentes de los Estados Parte, siempre que se cumpla con los porcentajes en los plazos establecidos en los párrafos anteriores. Asimismo, para efectos del registro y procedimiento de reconocimiento de registro en los Estados Parte, el certificado de buenas prácticas de manufactura emitido por la autoridad competente de Honduras, deberá cumplir con los porcentajes en los plazos establecidos en los párrafos anteriores.
3. Los laboratorios que demuestren en cualquier momento el cumplimiento del 100% de los puntos críticos, mayores y menores, establecidos en la Guía de Verificación del RTCA 11.03.42:07 Productos Farmacéuticos Medicamentos de Uso Humano. Buenas Prácticas de Manufactura para la Industria Farmacéutica, se les aplicará el procedimiento de reconocimiento del registro sanitario de productos farmacéuticos, aun cuando no haya entrado en vigencia dicho Reglamento o no hayan transcurrido los plazos de implementación indicados en el párrafo 2 de la presente resolución. Esto no afectará lo establecido en las Resoluciones 93-2002 (COMIECO-XXIV) y 99-2002 (COMIECO-XXV) (procedimiento ágil entre El Salvador, Guatemala, Honduras y Nicaragua) ni dará derecho a que laboratorios que no cumplan con el 100% de puntos críticos gocen de dicho procedimiento en países cuya legislación interna así lo exija. Al entrar en vigencia el RTCA 11.03.42:07 Productos Farmacéuticos Medicamentos de Uso Humano. Buenas Prácticas de Manufactura para la Industria Farmacéutica, los Anexos 1, 3 Y 7 de la Resolución 93-2002 (COMIECO-XXIV) quedarán sin efecto.
4. La presente Resolución entrará en vigencia el 25 de abril de 2016 y será publicada por los Estados Parte, sin embargo, el párrafo 3 de la misma entrará en vigor a partir del 12 de junio de 2014.
Tegucigalpa, Honduras, 25 de abril de 2014
(f) Fernando Ocampo Sánchez, Viceministro en representación de la Ministra de Comercio Exterior de Costa Rica. (f) José Armando Flores Alemán, Ministro de Economía de El Salvador. (f) María Luisa Flores Villagrán, Viceministra, en representación del Ministro de Economía de Guatemala. (f) Alden Rivera Montes, Secretario de Estado en el Despacho de Desarrollo Económico de Honduras. (f) Orlando Solórzano Delgadillo, Ministro de Fomento, Industria y Comercio de Nicaragua. (f) Diana Salazar, Viceministra, en representación del Ministro de Comercio e Industrias de Panamá.
REGLAMENTO TÉCNICO CENTROAMERICANO
RTCA 11.03.42:07
ANEXO DE LA RESOLUCIÓN No. 339-2014
(COMIECO-LXVII)
RTCA 11.03.42:07
REGLAMENTO TÉCNICO CENTROAMERICANO
PRODUCTOS FARMACÉUTICOS
MEDICAMENTOS DE USO HUMANO BUENAS PRÁCTICAS DE MANUFACTURA PARA LA INDUSTRIA FARMACÉUTICA
Correspondencia: Este reglamento tiene correspondencia con el informe 32 elaborado por el Comité de Expertos de la Organización Mundial de la Salud.
ICS 11.000 RTCA 11.03.42:07
Reglamento Técnico Centroamericano editada por:
· Ministerio de Economía, MINECO
· Organismo Salvadoreño de Reglamentación Técnica, OSARTEC
· Ministerio de Fomento, Industria y Comercio, MIFIC
· Secretaria de Industria y Comercio, SIC
· Ministerio de Economía, Industria y Comercio, MEIC
INFORME
Los respectivos Comités Técnicos de Normalización a través de los Entes de Normalización de los Estados Miembros que Integran la Región Centroamericana, y sus sucesores, son los organismos encargados de realizar el estudio o la adopción de los Reglamentos Técnicos. Están integrados por representantes de la Empresa Privada, Gobierno, Organismos de Protección al Consumidor y Académico Universitario.
Este documento fue aprobado como Reglamento Técnico Centroamericano RTCA 11.03 .42:07 PRODUCTOS FARMACÉUTICOS. MEDICAMENTOS DE USO HUMANO. BUENAS PRÁCTICAS DE MANUFACTURA PARA LA INDUSTRIA FARMACÉUTICA, por los Subgrupos de Medidas de Normalización y Medicamentos y Productos Afines de la Región Centroamericana. La oficialización de este Reglamento Técnico, conlleva la aprobación por el Consejo de Ministros de Integración Económica (COMIECO).
MIEMBROS PARTICIPANTES DEL COMITÉ
Por Guatemala:
Ministerio de Salud Pública y Asistencia Social
Por El Salvador:
Dirección Nacional de Medicamentos
Por Nicaragua:
Ministerio de Salud
Por Honduras:
Secretaria de Salud Pública
Por Costa Rica
Ministerio de Salud l1. OBJETO
El presente reglamento técnico establece los principios y directrices de las Buenas Prácticas de Manufactura que regulan todos los procedimientos involucrados en la manufactura de productos farmacéuticos a fin de asegurar la eficacia, seguridad y calidad de los mismos.
2. ÁMBITO DE APLICACIÓN
Es de aplicación a los laboratorios fabricantes de productos farmacéuticos en los países centroamericanos.
3. DOCUMENTOS A CONSULTAR
Para la adecuada interpretación y aplicación del presente reglamento se podrá consultar los siguientes documentos:
-Informe 32. Serie de informes técnicos 823, Comité de expertos de la OMS en especificaciones para las preparaciones Farmacéuticas Ginebra 1992
-Guía de inspección y auto inspección de Buenas Prácticas de Manufactura para la Industria Farmacéutica de los países centroamericanos.
4. RELACIÓN ENTRE OPERACIONES DE FABRICACIÓN Y LICENCIA SANITARIA PERMISO SANITARIO DE FUNCIONAMIENTO
El laboratorio fabricante velará porque todas las operaciones de fabricación se lleven a cabo de conformidad con la información aprobada en la Licencia Sanitaria o Permiso sanitario de funcionamiento otorgado por la Autoridad Reguladora del país.
5. DEFINICIONES
Para los efectos de este reglamento se establecen las siguientes definiciones:
5.1 Acondicionamiento: todas las operaciones, incluidos el llenado y el etiquetado, necesarias para convertir un producto a granel en producto terminado.
5.2 Agua calidad farmacéutica o purificada: es el agua tratada que se emplea como excipiente en la producción de preparaciones no parenterales y en otras aplicaciones farmacéuticas, tal como la limpieza de determinados equipos y componentes que entran en contacto con el producto no parenteral.
5.3 Agua para inyección: es el agua que se emplea como excipiente en las preparaciones parenterales y en otras preparaciones donde se debe controlar el contenido de endotoxinas, así como en otras aplicaciones farmacéuticas tal como para la limpieza de determinados equipos y componentes que entran en contacto con el producto parenteral.
5.4 Aprobado: condición que se establece cuando los resultados de las pruebas cumplen con las especificaciones establecidas, para que los componentes de la formulación y del empaque, productos en proceso, productos semielaborados y productos terminados puedan ser usados o distribuidos.
5.5 Aseguramiento de la calidad: es el conjunto de medidas y procedimientos definidos con el fin de asegurar que los productos elaborados sean de la calidad necesaria para el uso al que están destinados.
5.6 Área limpia: área que cuenta con un control definido del medio ambiente respecto a la contaminación con partículas, microorganismos, con instalaciones construidas y usadas de tal manera que se reduzca la introducción, generación y retención de contaminantes dentro del área.
5.7 Área aséptica: es el área limpia que cumple con los requisitos de aire grado A. establecido en el anexo A de este reglamento.
5.8 Área segregada: espacio separado físicamente y autónomo de otras áreas, destinadas a la producción de productos específicos. Debe tener una esclusa, un sistema de aire independiente, flujo de materiales y personal que evite la contaminación y la contaminación cruzada.
5.9 Autoridad reguladora: es la autoridad sanitaria de cada uno de los países centroamericanos.
5.10 Auto inspección: inspección efectuada por personal técnico calificado propio de la Empresa; que evalúa periódicamente el cumplimiento de las buenas prácticas de manufactura.
5.11 Auditoría: revisión de actividades específicas efectuada con la finalidad de establecer el cumplimiento de los procedimientos establecidos según buenas prácticas de manufactura.
5.12 Auditoría de la calidad: consiste en un examen y evaluación parcial o total del sistema de calidad con el propósito de cumplir y mejorarlo.
5.13 Buenas prácticas de laboratorio: conjunto de normas, procedimientos operativos y prácticas, para garantizar que los datos generados por un laboratorio de Control de Calidad son íntegros, confiables, reproducibles y de calidad.
5.14 Buenas prácticas de manufactura: conjunto de procedimientos y normas destinados a garantizar la producción uniforme de los lotes de productos farmacéuticos que cumplan las normas de calidad.
5.15 Calibración: proceso mediante el cual se establece si el desempeño de un instrumento satisface las especificaciones establecidas.
5.16 Calidad: naturaleza esencial de un producto y la totalidad de sus atributos y propiedades, las cuales determinan su idoneidad para los propósitos a los cuales se destina.
5.17 Calificación de equipo: acción de demostrar y documentar que el equipo o los sistemas auxiliares están correctamente instalados, trabajan y conducen realmente a los resultados esperados.
5.18 Certificado de análisis o informe de análisis: documento que especifica el resultado de las pruebas, de una muestra representativa del material evaluado.
5.19 Código o número de lote: combinación de letras, número o símbolos que sirven para la identificación de un lote y bajo el cual se amparan todos los documentos referentes a su manufactura y control.
5.20 Contaminación: es la presencia de entidades físicas, químicas y biológicas indeseables.
5.21 Contaminación cruzada: contaminación de un material o de un producto semielaborado o de un producto terminado con otro material o producto durante el proceso de producción.
5.22 Contrato a terceros: instrumento público debidamente autorizado en el que se acuerda la realización de la producción, el análisis o ambos en los productos farmacéuticos por otro laboratorio.
5.23 Control de calidad: sistema planificado de actividades cuyo propósito es verificar la calidad del producto.
5.24 Control de cambios: programa destinado a documentar cualquier cambio o variación, planeada o temporal, a un proceso validado de manufactura.
5.25 Control de proceso: pruebas, ensayos y mediciones efectuadas durante la elaboración de un producto, incluyendo su acondicionamiento destinado para asegurar que el producto resultante cumple con las especificaciones.
5.26 Cuarentena: situación de aislamiento de materiales tales como materias primas, material de acondicionamiento, productos semielaborados, a granel o terminados pendientes del dictamen del departamento de control de calidad para su aprobación o rechazo.
5.27 Concentración: es la cantidad de principio activo presente en el medicamento reportada en el sistema de unidades de medición definidas internacionalmente. (SI)
5.28 Devolución: retorno de un medicamento al fabricante o distribuidor.
5.29 Eficacia: capacidad de un medicamento para producir los efectos terapéuticos propuestos.
5.30 Envase primario o empaque primario: es todo recipiente que tiene contacto directo con el producto, con la misión específica de protegerlo de su deterioro, contaminación o adulteración y facilitar su manipulación.
5.31 Envase secundario o empaque secundario: envase definitivo de distribución y comercialización o material de empaque dentro del cual se coloca el envase primario que contiene al producto.
5.32 Esclusa: un lugar cerrado, con dos puertas, que se interpone entre dos o más ambientes con diferentes requerimientos de limpieza.
5.33 Especificaciones: Descripción de los requisitos que debe satisfacer el material inicial, el material de empaque y los productos intermedios, a granel y terminados. Dichos requisitos incluyen propiedades físicas, químicas y de ser posible biológicas.
5.34 Estabilidad: capacidad que tiene un producto, de mantener por determinado tiempo sus propiedades originales, dentro de las especificaciones establecidas.
5.35 Estándar de referencia primario: aquella sustancia que ha demostrado, a través de una serie de análisis, ser un material de alta pureza obtenido de una fuente oficial reconocida.
5.36 Estándar de referencia secundario: sustancia de calidad y pureza establecida, comparada con un estándar de referencia primario, usado como estándar de referencia para análisis rutinario del laboratorio.
5.37 Experto: persona que debe poseer educación científica y experiencia práctica que le permitan tener criterio profesional independiente, basado en la aplicación de principios científicos a los problemas prácticos que se presenten en la fabricación y control de la calidad de los productos farmacéuticos.
5.38 Fabricación o manufactura: todas las operaciones involucradas en la compra de materiales y productos, producción, control de calidad, aprobación, almacenamiento, distribución del producto terminado y los controles relacionados.
5.39 Fórmula maestra: documento en el cual se establecen los materiales de inicio y las cantidades respectivas que serán usadas en la fabricación de un medicamento, incluye además una descripción de las operaciones de producción y los detalles de los controles específicos que se emplearán durante el proceso.
5.40 Fecha de fabricación o fecha de manufactura: fecha de cada lote de producto, la cual se refiere al mes y año en que se llevó a cabo la fabricación del producto.
5.41 Fecha de expiración o fecha de vencimiento: fecha colocada en el material de empaque primario y secundario de un producto por lote fabricado, para indicar la fecha hasta la cual se espera que el producto satisfaga las especificaciones de calidad.
5.42 Forma farmacéutica: es la forma física que se le da a un medicamento, la cual facilita la dosificación del o de los principios activos para que puedan ejercer su acción en el lugar y tiempo.
5.43 Garantía de calidad: ver aseguramiento de calidad.
5.44 Identidad: presencia del ingrediente activo en un producto farmacéutico, según lo indique su especificación.
5.45 Laboratorio fabricante: entidad autorizada con instalaciones diseñadas, para realizar todas las operaciones que involucran la fabricación de productos farmacéuticos.
5.46 Libros oficiales: aquellos reconocidos por los países centroamericanos mediante disposiciones reglamentarias vigentes.
5.47 Licencia sanitaria o permiso sanitario de funcionamiento: documento emitido por la autoridad reguladora que autoriza al laboratorio fabricante para la manufactura de medicamentos previo cumplimiento de los requisitos establecidos.
5.48 Limpieza: es un procedimiento que se aplica para remover la suciedad y residuos.
5.49 Lote: cantidad de producto que se fabrica en un ciclo de producción. La característica esencial del lote es su homogeneidad.
5.50 Materia prima: toda sustancia de calidad definida empleada en la producción de un medicamento, con exclusión de los materiales de acondicionamiento.
5.51 Materiales: toda materia prima, material de empaque o acondicionamiento que es empleado en la fabricación de un producto.
5.52 Material de empaque o de acondicionamiento: cualquier material empleado en el acondicionamiento de medicamentos. El material de acondicionamiento se clasifica en primario o secundario según que esté o no en contacto directo con el producto.
5.53 Medicamento: sustancia simple o compuesta, natural, sintética o mezcla de ellas, con forma farmacéutica definida empleada para diagnosticar, tratar, prevenir enfermedades o modificar una función fisiológica de los seres humanos.
5.54 Muestra: parte o porción finita representativa de un material, un lote de producción, o de medicamentos almacenados, transportados o en uso que se someten a análisis a efecto de verificar las características de calidad o su adecuación para el uso.
5.55 Muestra de retención: cantidad de unidades representativa de cada lote de producto terminado, materia prima o material de envase, almacenada por un período de tiempo establecido y en cantidad suficiente para repetir el análisis completo.
5.56 Muestreo: procedimiento establecido para realizar la toma de una muestra homogénea.
5.57 Orden de producción: documento en el cual se registra la fórmula farmacéutica, las cantidades de cada uno de los ingredientes y autoriza su dispensación para la producción, de acuerdo a las instrucciones contenidas en la fórmula maestra.
5.58 Orden de envasado y empaque: documento que especifica las cantidades de material de envase y empaque que son utilizadas en el acondicionamiento de un lote, incluye una descripción de los procedimientos y precauciones, así como los controles durante el proceso.
5.59 Política de calidad: conjunto de directrices y objetivos de una organización con respecto a la calidad, expresados de manera formal por la alta gerencia.
5.60 Potencia: contenido de principio activo o fármaco presente en un producto farmacéutico o en una unidad de dosificación.
5.61 Principio activo: sustancia dotada de un efecto farmacológico específico o que, sin poseer actividad, al ser administrado en el organismo la adquiere luego que sufren cambios en su estructura química.
5.62 Procedimiento: descripción de las operaciones que deben realizarse, las precauciones que deben tomarse y las medidas que deben aplicarse relacionadas directa o indirectamente con la fabricación de un medicamento.
5.63 Procedimiento operativo estándar: procedimiento escrito autorizado que contiene instrucciones para realizar operaciones que son específicas para un producto o material determinado.
5.64 Proceso aséptico: aquel según el cual se extreman las medidas para evitar la contaminación microbiana de los productos, equipos y componentes que han sido previamente esterilizados o sanitizados.
5.65 Producción: todas las operaciones involucradas en la elaboración de un producto farmacéutico, desde la recepción de los materiales, hasta la obtención del producto terminado.
5.66 Producto a granel: producto que ha pasado por todas las fases de producción excepto el acondicionamiento final.
5.67 Producto intermedio o semielaborado: es aquel que se encuentra en alguna de las fases intermedias de su proceso que antecede a la forma farmacéutica definitiva.
5.68 Producto terminado: producto que ha pasado por todas las fases de producción.
5.69 Protocolo de validación: documento en el que se describe las actividades que se realizan en una validación, incluidos en los criterios de aceptación para la aprobación de un proceso de fabricación o parte del mismo.
5.70 Prueba de identidad: prueba diseñada para mostrar de manera inequívoca que las muestras examinadas contienen el principio o principios activos rotulados.
5.71 Pureza: grado en que una entidad química o biológica está presente en otra.
5.72 Registro de producción: documento que recopila la historia de cada lote de producto y todas las demás circunstancias importantes que pueden afectar a la calidad del producto final.
5.73 Re análisis: análisis requerido para verificar que la materia prima, material de acondicionamiento o producto terminado, cumple las especificaciones con las que fue aprobado originalmente.
5.74 Reproceso: procedimiento que se aplica a un producto intermedio o producto terminado que no haya salido de la planta farmacéutica, para lograr que alcance los criterios de aceptación.
5.75 Rendimiento: comparación, con un margen de tolerancia por las variaciones normales entre la cantidad del producto o materiales teóricamente producidos o empleados y la cantidad realmente producida o empleada.
5.76 Sanitización: Control del desarrollo y reproducción de microorganismos patógenos del medio ambiente, mediante métodos físicos y químicos.
5.77 Validación: acción documentada que demuestra que un procedimiento, proceso, equipo, material, actividad o sistema conducen a los resultados previstos.
6. DE LOS REQUISITOS
6.1 De la autorización de funcionamiento.
Los laboratorios fabricantes de productos farmacéuticos deben tener permiso sanitario de funcionamiento o licencia sanitaria correspondiente, de acuerdo a los requisitos legales establecidos por la Autoridad Reguladora de cada país centroamericano.
7. ORGANIZACIÓN Y PERSONAL
7.1 Organización
La organización de la empresa debe estar documentada en un organigrama general que indique claramente la estructura jerárquica y en organigramas específicos de los departamentos. Los cuales deben estar vigentes y firmados por las personas responsables.
7.1.2 Descripción de puestos.
Debe existir una descripción escrita de las funciones y responsabilidades de cada puesto incluido en el organigrama y se especificará el grado académico y las habilidades que el personal debe tener para ocuparlos.
7.1.3 Del Director Técnico o Regente Farmacéutico.
El laboratorio fabricante de productos farmacéuticos debe tener una Dirección Técnica o Regencia Farmacéutica la cual estará a cargo de un profesional farmacéutico, durante el horario de su funcionamiento, cuyo puesto estará incluido dentro del organigrama general. Esta dirección es responsable de cuanto afecte la eficacia, seguridad y calidad de los medicamentos que se formulen, elaboren, manipulen, almacenen y distribuyan, así como el cumplimiento de las disposiciones legales y reglamentarias que demande la operación del establecimiento que regenta. Es solidario en esta responsabilidad, el representante legal de la empresa. En casos de jornadas continuas o extraordinarias el regente debe garantizar los mecanismos de supervisión de acuerdo a la legislación nacional de cada Estado Parte.
7.2 Personal
7.2.1 Del personal.
El laboratorio fabricante debe disponer de personal con la calificación y/o experiencia práctica necesaria. Las responsabilidades encargadas a cada persona no deben ser tan numerosas como para constituir un riesgo para la calidad.
7.2.2 De los responsables de las áreas técnicas.
Los profesionales farmacéuticos o profesionales calificados, responsables de las unidades de investigación y desarrollo, producción, control y garantía de la calidad deben tener experiencia técnica para el puesto que ocupen.
7.2.3 De la calificación del personal.
Toda persona que labore en la industria farmacéutica, debe tener preparación académica, capacitación y experiencia o una combinación de esas condiciones, para ocupar el puesto al que se le asigne.
7.3 Responsabilidades del personal.
7.3.1 De las responsabilidades de la dirección de producción. Las responsabilidades de la dirección de producción son:
a. Asegurar que los productos se fabriquen y almacenen en concordancia con la documentación aprobada, a fin de obtener la calidad prevista.
b. Aprobar los documentos maestros relacionados con las operaciones de producción, incluyendo los controles durante el proceso y asegurar su estricto cumplimiento.
c. Garantizar que la orden de producción esté completa y firmada por las personas designadas, antes de que se pongan a disposición del Departamento asignado.
d. Vigilar el mantenimiento del Departamento en general, instalaciones y equipo.
e. Garantizar que los procesos de producción, se realizan bajo los parámetros definidos.
f. Autorizar los procedimientos del Departamento de Producción y verificar que se cumplan
g. Asegurar que se lleve a cabo la capacitación inicial y continua del personal de producción y que dicha capacitación se adapte a las necesidades.
h. Otras funciones inherentes al puesto.
7.3.2 De las responsabilidades de la dirección de control de calidad.
Las responsabilidades de la Dirección de Control de Calidad son:
a. Aprobar o rechazar, según procede, las materias primas, materiales de envase y empaque, producto intermedio, a granel y terminado.
b. Revisar que toda la documentación de un lote de producto que se ha finalizado, esté completa, la cual también puede ser responsabilidad de garantía de calidad.
c. Aprobar las instrucciones de muestreo, métodos de análisis y otros procedimientos de Control de Calidad y verificar las especificaciones.
d. Aprobar y controlar los análisis llevados a cabo por contrato a terceros.
e. Vigilar el mantenimiento del Departamento, las instalaciones y los equipos.
f. Verificar que se efectúen las validaciones correspondientes a los procedimientos analíticos y de los equipos de control.
g. Asegurar que se lleve a cabo la capacitación inicial y continua del personal de Control de Calidad y que dicha capacitación se adapte a las necesidades.
h. Otras funciones propias del departamento de control de calidad.
7.3.3 De las responsabilidades compartidas de la dirección de producción y de control de calidad.
Los responsables de Producción y Control de Calidad deben compartir o ejercer responsabilidades relativas a la calidad, las cuales son las siguientes:
a. Autorizar los procedimientos escritos y otros documentos, incluyendo sus modificaciones.
b. Vigilar y controlar las áreas de producción.
c. Vigilar la higiene de las instalaciones de las áreas productivas.
d. Validar los procesos, calificación y calibración de los equipos e instrumentos. e. Capacitar.
f. Participar en la selección, evaluación (aprobación) y control los proveedores de materiales, de equipo y otros, involucrados en el proceso de producción.
g. Aprobar y controlar la fabricación por terceros.
h. Establecer y controlar las condiciones de almacenamiento de materiales y productos.
i. Conservar la documentación.
j. Vigilar el cumplimiento de las Buenas Prácticas de Manufactura.
k. Inspeccionar, investigar y muestrear con el fin de controlar los factores que puedan afectar a la calidad.
7.4 Capacitación
7.4.1 De la inducción.
Todo empleado de nuevo ingreso, debe recibir capacitación inductiva. La asistencia a esta capacitación debe quedar documentada. La capacitación debe ser general en las Buenas Prácticas de Manufactura y especifica de acuerdo a las funciones y atribuciones asignadas, antes de ingresar a su puesto de trabajo. El personal administrativo debe recibir inducción general en Buenas prácticas de manufactura.
7.4.2 De la capacitación continúa.
La capacitación debe ser continua y acorde con las funciones propias del puesto, de igual manera con las regulaciones y procedimientos escritos de las Buenas Prácticas de Manufactura en todo aquello relacionado con el puesto que ocupa y debe quedar documentada.
7.4.3 De la planificación.
La capacitación en Buenas Prácticas de Manufactura, debe realizarse, de acuerdo a una planificación establecida y aprobada, por las personas responsables en la empresa y debe garantizar el conocimiento de dichas prácticas. Esta debe efectuarse como mínimo dos veces al año.
7.4.4 De las evaluaciones.
Se debe realizar una evaluación del programa de capacitación, su ejecución y resultados de acuerdo a una planificación establecida, quedando debidamente documentada.
7.4.5 De la capacitación específica.
El personal que labore en áreas estériles o áreas donde se manejen sustancias activas, tóxicas, infecciosas o sensibilizantes (â-lactámicos, citostáticos y hormonales), debe recibir capacitación específica, la cual debe evaluarse en forma periódica. Se deben mantener los registros.
7.4.6 De los visitantes.
Se restringirá el acceso de los visitantes o del personal no específicamente capacitado a las áreas de producción y control de calidad. Si esto fuera inevitable, se les dará información previa, especialmente sobre higiene personal y uso de ropa protectora. Dicho ingreso debe ser objeto de supervisión. Esta restricción se divulgará por medio de rótulo colocado visiblemente antes del ingreso.
7.5 Salud e higiene del personal
7.5.1 De la salud del personal.
Todo el personal al ser contratado y durante el tiempo de empleo debe someterse a exámenes médicos, de acuerdo a las áreas de desempeño, para asegurar que sus condiciones de salud no afectan la calidad del producto que se está fabricando. El laboratorio fabricante será el responsable que el personal presente anualmente o de acuerdo a la legislación de cada país la certificación médica o su equivalente, garantizando que no padece de enfermedades infectocontagiosas.
7.5.2 Del control de la salud del personal que labora en áreas especiales.
El personal que labora en áreas de producción de â-Lactámicos, sustancias tóxicas, organismos sumamente activos, citostáticos y hormonales, deben poseer un control de salud, de manera que, garantice la integridad física de los mismos.
7.5.3 De los aspectos relacionados con las condiciones de salud.
No debe intervenir en la producción de medicamentos ninguna persona afectada por una enfermedad infecciosa o que tenga heridas abiertas en la superficie del cuerpo. El fabricante debe instruir al personal para que informe acerca de todos los estados de salud que puedan influir negativamente en los productos.
7.5.4 De los procedimientos de higiene personal.
Los procedimientos relacionados con la higiene personal incluyendo el uso de ropas protectoras, se aplican a todas las personas que ingresan a las áreas de producción, incluyendo empleados temporales, permanentes y visitantes.
7.5.5 De la protección del personal.
El personal dedicado a la producción, que esté en contacto directo con los productos debe usar uniforme de manga larga, limpio, sin bolsas en la parte superior de la vestimenta confortable y confeccionada con un material que no desprenda partículas, botones escondidos; y protección como gorros que cubran la totalidad del cabello, mascarillas, guantes y zapatos especiales (cerrados, suela antideslizante). Los requerimientos de indumentaria para cada tipo de área, se deben definir por escrito.
7.5.6 De las prohibiciones en las áreas de producción, almacenamiento, y Control de Calidad.
Se prohíbe comer, beber, fumar, masticar, así como guardar comida, bebida, cigarrillos, medicamentos personales en las áreas de producción y cualquier otra área donde esas actividades puedan influir negativamente en la calidad de los productos. El personal no debe usar maquillaje, joyas, relojes, teléfonos celulares, radio-localizadores, ni ningún instrumento ajeno al uniforme, en áreas de riesgo para el producto. No debe llevar barba o bigote al descubierto, durante la jornada de trabajo en los procesos de dispensado, producción y subdivisión. El uniforme de trabajo debe ser usado exclusivamente en las áreas para las que fue diseñado, según los procedimientos escritos que lo definen. Esta prohibición debe indicarse por medio de rótulos visibles colocados previo al ingreso al área de producción.
7.5.7 De los hábitos higiénicos del personal.
Todas las personas involucradas en el proceso de fabricación deben tener buenos hábitos higiénicos. Será obligación del personal lavarse las manos antes de ingresar a las áreas de producción, especialmente después de utilizar los servicios sanitarios y después de comer. Se deben colocar rótulos visibles referentes a esta obligación.
7.5.8 De los controles microbiológicos.
El laboratorio debe realizarle al personal los controles microbiológicos de manos y otros, de acuerdo a las áreas de desempeño, a un programa y procedimiento establecido.
7.5.9 De los primeros auxilios.
La empresa debe contar con un botiquín y área destinada a primeros auxilios.
8. EDIFICIOS E INSTALACIONES
8.1 Ubicación, diseño y características de la construcción
8.1.1 De las generalidades.
Las instalaciones deben diseñarse, construirse, remodelarse y mantenerse de forma conveniente a las operaciones que deben realizarse. Su disposición y diseño deben tender a minimizar el riesgo de errores y a permitir limpieza y mantenimiento efectivo para evitar la contaminación cruzada, la acumulación de polvo o suciedad y, en general, cualquier efecto negativo sobre la calidad de los productos.
8.1.2 De los planos y diagramas
El laboratorio fabricante debe contar como mínimo con los siguientes planos y diagramas actualizados:
Planos de construcción y remodelaciones.
Plano de distribución de áreas.
Diagrama de flujo de personal.
Diagrama de flujo de materiales.
Diagrama de flujo de procesos.
Plano de servicios (aire acondicionado, aire comprimido, aguas, desagües, aguas servidas, aguas negras, electricidad, vapor, vapor puro y gases.
Plano de evacuación del personal en caso de emergencia y plano de ubicación de salidas de emergencia.
Diagrama del sistema de tratamiento de aguas para la producción.
8.1.3 De la ubicación.
Las instalaciones deben estar ubicadas en un ambiente tal, que, consideradas en conjunto con las medidas destinadas a proteger las operaciones de fabricación, ofrezcan el mínimo riesgo de contaminar materiales o productos.
8.1.4 Del mantenimiento.
El laboratorio fabricante debe ser mantenido en excelentes condiciones de uso. Deben existir procedimientos y registros de los mantenimientos realizados periódicamente a las instalaciones y edificios.
8.1.5 De la protección.
Las instalaciones deben diseñarse y equiparse de tal forma que ofrezcan la máxima protección contra el ingreso de animales.
8.1.6 Del flujo.
El flujo de los materiales y del personal a través del laboratorio fabricante, debe estar diseñado de tal manera que no permita confusión, contaminación ni errores. Las áreas de acceso restringido deben estar debidamente delimitadas e identificadas.
8.1.7 De las áreas de paso.
Las áreas de producción, almacenamiento y control de calidad no deben utilizarse como lugar de paso por el personal que no trabaje en las mismas
8.1.8 De las instalaciones especiales.
Las operaciones para la fabricación de â-lactámicos, citostáticos, productos biológicos y productos no farmacéuticos, deben realizarse en edificios separados y autónomos o instalaciones independientes y autónomas, en ambos casos deben demostrar que no hay contaminación cruzada y contaminación al exterior.
Los productos tales como hormonas, homeopáticos y productos naturales con indicación terapéutica deben fabricarse en áreas segregadas, pudiéndose trabajar por campaña, validando sus procesos de limpieza y producción.
Nota
Edificios separados y autónomas o instalaciones independientes y autónomas son aquellas con las siguientes características: entradas y salidas independientes para el personal y materiales, con sus respectivas áreas de vestidores, servicios sanitarios, almacenamiento de materias primas, dispensado, elaboración, envasado y empaque, equipo y lavado de equipos, sistemas de agua residual, sistema de aire, documentación, personal, control de calidad en proceso, comedor, lavandería.
8.1.9 De las condiciones ambientales.
Las condiciones de iluminación, temperatura, humedad y ventilación no deben influir negativamente, directa o indirectamente en los productos durante su producción y almacenamiento.
8.1.10 De la ubicación de equipos y materiales.
Las áreas de trabajo y almacenamiento deben permitir la ubicación lógica de los equipos y materiales de tal forma que se reduzca al mínimo el riesgo de confusión entre los distintos productos y sus componentes, se evite la contaminación cruzada, se reduzca el riesgo de omisión y aplicación errónea de cualquiera de las operaciones de producción o control.
8.1.11 Exclusividad de las áreas.
Las áreas deben ser exclusivas para el uso previsto y no estar invadidas por materiales extraños.
8.1.12 De la ubicación de los servicios.
Las tuberías, artefactos lumínicos, puntos de ventilación y otros servicios deben ser diseñados y ubicados de tal forma que no causen dificultades en la limpieza.
8.1.13 De la seguridad industrial.
Se deben de disponer de equipamiento para el cumplimiento de la seguridad industrial, según la normativa de cada estado parte.
8.1.14 De los drenajes o desagües.
Los drenajes deben ser diseñados y ubicados de manera que no permita la contracorriente. Deben tener tapas tipo sanitario.
8.2 Almacenes
8.2.1 De los almacenes
Las áreas de almacenamiento deben tener suficiente capacidad para permitir el almacenamiento ordenado de diversas categorías de materiales y productos: materias primas, materiales de envase y empaque, productos intermedios, a granel, terminados, productos en cuarentena, aprobados, rechazados, devueltos o retirados.
8.2.2 De las características de pisos, paredes y techos
Los pisos, paredes, techos, no deben afectar la calidad de los materiales y productos que se almacenan y estos deben ser de fácil limpieza.
8.2.3 De las condiciones ambientales.
Las áreas de almacenamiento deben diseñarse o adaptarse para asegurar las buenas condiciones de almacenamiento. Deben mantenerse limpias, ordenadas, a temperatura y humedad de acuerdo a las especificaciones de los materiales y productos. En los casos que se requiera condiciones especiales de temperatura y humedad estas deben establecerse, controlarse y vigilarse.
8.2.4 De las características del área de recepción y despacho.
En los lugares de recepción y despacho, los productos y materiales deben estar protegidos de las condiciones ambientales. Las áreas de recepción deben diseñarse y equiparse de tal forma que los contenedores de materiales puedan limpiarse, si fuere necesario, antes de su almacenamiento.
8.2.5 Del área de cuarentena.
Las áreas donde se almacenan materiales y productos; sometidos a cuarentena deben estar claramente definidas y marcadas; el acceso a las mismas debe limitarse al personal autorizado. Todo sistema destinado a sustituir el área de cuarentena debe ofrecer condiciones equivalentes de seguridad.
8.2.6 Del área de muestreo. Debe existir área de muestreo para las materias primas que esté separada de las demás Debe hacerse de tal forma que se impida la contaminación cruzada. El muestreo puede efectuarse en el área de pesaje o dispensado.
8.2.7 Del almacenamiento de productos rechazados, retirados y devueltos.
El almacenamiento de materiales o productos rechazados, retirados del mercado o devueltos debe efectuarse en áreas separadas, identificadas, de acceso restringido, bajo llave y documentado.
8.2.8 De la rotación de los materiales.
Los materiales deben almacenarse de manera que faciliten la rotación de los mismos según la regla "primero que vence primero que sale"
8.2.9 Del uso de tarimas o estanterías.
Los materiales y productos deben identificarse y colocarse sobre tarimas o estanterías que permitan la limpieza e inspección.
8.2.10 Del almacenamiento de estupefacientes y psicotrópicos.
Deben existir áreas separadas, bajo llave, identificadas y de acceso restringido para almacenar materias primas y productos terminados que contengan psicotrópicos y estupefacientes.
8.2.11 Del almacenamiento de Sustancias Inflamables.
Debe existir un área destinada al almacenamiento de sustancias inflamables, la cual debe ser ventilada y con equipo de seguridad contra incendios o explosiones, ubicada en áreas alejadas de las otras instalaciones.
8.2.12 Del almacenamiento de material impreso.
Debe existir un área separada, identificada y de acceso restringido para almacenar dichos materiales (etiquetas, estuches, insertos y envases impresos).
8.3 Área de dispensado de materias primas.
8.3.1 De las condiciones.
Debe existir un área específica, identificada como "área restringida", para llevar a cabo las operaciones de dispensación. Las paredes, pisos y techos deben ser lisos y con curvas sanitarias. Esta debe ser independiente, cerrada, limpia, iluminada y en condiciones controladas de temperatura y humedad (cuando se requiera). Esta área debe contar con sistemas de aire independiente de inyección y extracción, con diferencial de presión para evitar las contaminaciones y proteger al producto y al personal. El diferencial de presión debe ser controlado y registrado.
8.3.2 De la ubicación de los equipos sensibles.
El soporte donde se encuentren las balanzas y otros equipos sensibles debe ser capaz de contrarrestar las vibraciones que afectan su buen funcionamiento.
8.3.3 Del equipo.
Debe estar equipada con balanzas y material volumétrico calibrados de acuerdo al rango de medida de los materiales a dispensar.
8.3.4 Del área en tránsito
Debe ubicarse adyacente al área de dispensado, un área delimitada e identificada en la que se colocarán las materias primas que serán pesadas y las materias primas dispensadas que serán utilizadas en la producción.
8.4 Área de producción
8.4.1 Del diseño de las áreas.
Se debe disponer de áreas que posean el tamaño, diseño y servicios (ventilación, agua, luz y otros que se requieran) para efectuar los procesos de producción que correspondan.
8.4.2 De las condiciones de las áreas.
Las áreas deben tener las siguientes condiciones:
a. Estar identificadas y separadas para la producción de sólidos, líquidos y semisólidos, tener paredes, pisos y techos lisos, con curvas sanitarias, sin grietas ni fisuras, no utilizar madera, no deben liberar partículas y deben permitir su limpieza y sanitización.
b. Las tuberías y puntos de ventilación deben ser de materiales que permitan su fácil limpieza y estar correctamente ubicados.
c. Toma de gases y fluidos identificados. d. Ventanas de vidrio fijo, lámparas y difusores, lisos y empotrados que sean de fácil limpieza y evite la acumulación de polvo.
e. Tener inyección y extracción de aire, con equipo para el control de temperatura, humedad y presión de acuerdo a los requerimientos o especificaciones de cada área.
f. Las áreas de producción no deben utilizarse como áreas de paso.
g. Estar libre de materiales y equipo que no estén involucrados en el proceso.
8.4.3 Área de acondicionamiento para empaque primario
Deben existir áreas de acondicionamiento para empaque primario que cumplan con las condiciones establecidas en el artículo 8.4.2 para este fin.
8.4.4 Del área de lavado.
Debe existir un área exclusiva destinada al lavado de equipos móviles, recipientes y utensilios. Esta área debe mantenerse en buenas condiciones de orden, limpieza, contar con curvas sanitarias y servicios para el trabajo que allí se ejecuta.
8.4.5 Área de equipo limpio.
Debe existir un área identificada, limpia, ordenada y separada para colocar equipo limpio que no se esté utilizando.
8.5 Áreas de acondicionamiento para empaque secundario
8.5.1 Del área de acondicionamiento para empaque secundario.
Las áreas de empaque o acondicionamiento para empaque secundario, deben tener un tamaño de acuerdo a su capacidad y línea de producción, con el fin de evitar confusiones, y manteniendo el orden y limpieza.
8.5.2 De las condiciones del área.
El área debe tener las siguientes condiciones:
a. Estar separada e identificada, con paredes, pisos y techos lisos, sin grietas ni fisuras, no utilizar madera y deben permitir su fácil limpieza.
b. Toma de gases y fluidos identificados.
c. Ventanas fijas, lámparas, difusores lisos y empotrados que sean de fácil limpieza y eviten la acumulación de polvo.
d. Ventilación e iluminación que asegure condiciones confortables al personal y no afecte negativamente la calidad del producto.
8.6 Área de control de calidad
8.6.1 Del área de control de calidad.
El área de control de calidad debe estar identificada y separada de las áreas de producción.
8.6.2 Del diseño del área de control de calidad.
Los laboratorios de control de calidad deben:
a. Diseñarse de acuerdo a las operaciones que se realicen, contando con las siguientes áreas, tales como: fisicoquímica, instrumental, microbiológica, lavado de cristalería y utensilios.
b. Tener paredes lisas que faciliten su limpieza.
c. Disponer de suficiente espacio para evitar confusiones y contaminación cruzada
d. Disponer de áreas de almacenamiento en condiciones para las muestras, reactivos, patrones de referencia, archivos, bibliografía y documentación.
e. Contar con los requerimientos de seguridad ocupacional (tales como: duchas, campana extractoras, lava ojos y cualquier otro que se requiera).
8.6.3 Del área para instrumentos sensibles. El área de instrumental debe estar diseñada para proteger el equipo e instrumentos sensibles del efecto de las vibraciones, interferencias eléctricas, humedad, temperatura.
8.6.4 Del área de microbiología. El área de microbiología debe estar separada de las otras áreas, contando con un área exclusiva para el proceso de siembra de productos estériles y no estériles, con acabados de fácil limpieza (curvas sanitarias), sistemas de aire independiente o flujo laminar, paredes, techos, pisos lisos, lámparas con difusor liso, mesa de trabajo lisa y ventanas de vidrio fijo.
8.7 Áreas auxiliares
8.7.1 De los vestidores y servicios sanitarios. Los vestidores y servicios sanitarios deben tener las siguientes condiciones:
a. Identificados correctamente.
b. Un número de servicios sanitarios para hombres y para mujeres de acuerdo al número de trabajadores.
c. Mantenerse limpios y ordenados.
d. Deben existir procedimientos y registros para la limpieza y sanitización.
e. Los servicios sanitarios deben estar accesibles a las áreas de trabajo y no deben comunicarse directamente con las áreas de producción.
f. Deben contar con lavamanos y duchas provistas de agua fría y caliente donde se requiera.
g. Disponer de espejos, toallas de papel o secador eléctrico de manos, jaboneras con jabón líquido desinfectante y papel higiénico.
h. Los vestidores deben estar separados de los servicios sanitarios por una pared.
i. Casilleros, zapateras y las bancas necesarias, y no utilizar madera.
j. Rótulos o letreros que enfaticen la higiene personal.
k. Se prohíbe mantener, guardar, preparar y consumir alimentos en esta área.
8.7.2 Del área de comedor.
Deben contar con un área para el comedor, debidamente acondicionada e identificada, en buenas condiciones de orden y limpieza para prevenir la proliferación de insectos y roedores, la cual debe estar separada de las demás áreas.
8.7.3 Del área de lavandería.
Debe contar con áreas separadas y exclusivas para el lavado y preparación de los uniformes utilizados por el personal. Se deben establecer procedimientos escritos para llevar a cabo esta labor.
8.7.4 Del área de mantenimiento y equipo sin uso.
Deben existir áreas separadas a las áreas de producción destinadas al mantenimiento de equipo y almacenamiento de herramientas y repuestos; otra destinada para almacenar el equipo obsoleto o en mal estado, que no interviene en los procesos.
8.7.5 Del área de investigación y desarrollo
Todo laboratorio fabricante debe contar con un área destinada para investigación y desarrollo de sus productos. Debe de contar con paredes lisas que faciliten su limpieza y el equipo necesario para las operaciones que se realizan.
9. EQUIPO
9.1 De las Generalidades
9.1.1 De las condiciones/Generalidades
Los equipos deben diseñarse, construirse y ubicarse de forma tal que facilite las operaciones relacionadas con su limpieza, mantenimiento y uso, con el fin de evitar la contaminación cruzada y todo aquello que pueda influir negativamente en la calidad de los productos. Debe contar con un código de identificación único.
9.1.2 De las instrucciones de operación.
Todo equipo empleado en la producción, control de calidad, empaque y almacenaje, debe contar con un procedimiento en el cual se especifiquen en forma clara las instrucciones y precauciones para su operación.
9.1.3 Reparación del equipo.
Las operaciones de reparación y mantenimiento no deben presentar ningún riesgo para la calidad de los productos.
9.1.4 De la limpieza y mantenimiento de equipo.
La limpieza y mantenimiento del equipo incluyendo utensilios debe realizarse de acuerdo a procedimientos escritos según programa establecido, conservando el registro de los mismos. Se permitirá el lavado, sanitizado y esterilizado cuando aplique en el área de producción cuando se utilizan equipos diseñados para realizar estas tareas automáticamente, es decir, cuando se utilizan los sistemas de limpieza, sanitización o esterilización en el Jugar (CIP o SIP por sus siglas en inglés), o en el caso de que los equipos sean muy pesados para poder ser movilizados.
9.1.5 De la identificación del equipo limpio.
La limpieza debe registrarse con una etiqueta que indique lo siguiente:
a. Nombre del equipo.
b. Fecha cuando fue realizada la limpieza.
c. Nombre y código o número de lote del último producto fabricado.
d. Nombre y código o número de Jote del producto a fabricar (cuando aplique).
e. Nombre o firma del operario que realizó la limpieza y de quien la verificó.
9.1.6 De los registros de mantenimiento del equipo.
Debe mantenerse registros escritos del mantenimiento preventivo y correctivo.
9.1.7 De la superficie de los equipos.
Las superficies de los equipos que tienen contacto directo con las materias primas o productos en proceso, deben ser de acero inoxidable de acuerdo a su uso, o si se requiere de otros materiales, estos no deben ser reactivos, aditivos o absorbentes para asegurar que no se alterará la calidad y seguridad de los productos. Se debe evitar el contacto entre el producto y las sustancias requeridas para el buen funcionamiento del equipo.
9.1.8 De los soportes.
Los equipos que requieran una base para su soporte, ésta debe de ser de acero inoxidable u otro material que no contamine.
9.2 Calibración
9.2.1 Generalidades.
Se debe realizar la calibración de instrumentos de medición, y dispositivos de registro o cualquier otro, que garantice la calidad de los productos. Esta calibración debe ser a intervalos convenientes y establecidos de acuerdo con un programa escrito que contenga como mínimo: frecuencias, límites de exactitud, precisión y previsiones para acciones preventivas y correctivas. Los instrumentos que no cumplan con las especificaciones establecidas no deben usarse. Deben mantenerse registros escritos de esas inspecciones, verificaciones y calibraciones.
9.2.2 De los patrones. Las calibraciones para cada equipo y dispositivos de seguimiento y medición deben realizarse usando patrones de referencia certificados. Se debe contar con un sistema de calibración periódica y verificación externa de los equipos.
9.3 Sistema de agua
9.3.1 Suministro. Debe tener un suministro de agua potable que Je permita satisfacer sus necesidades.
9.3.2 De la calidad del agua. Todo laboratorio de productos farmacéuticos debe contar con un sistema de tratamiento de agua que le permita obtener agua de calidad que cumpla con las especificaciones de los libros oficiales para Ja producción de sus productos.
9.3.3 Del monitoreo de los sistemas de suministro de agua.
Los sistemas de suministro, tratamiento de agua y el agua tratada, deben ser monitoreados. Deben mantenerse registros del monitoreo y de las acciones realizadas.
9.3.4 Del mantenimiento del sistema.
El sistema de tratamiento de agua debe estar sujeto a mantenimiento planificado y monitoreo.
9.3.5 De las especificaciones.
Para la producción de los productos y el enjuague final en la limpieza de los recipientes y equipos, se debe usar agua que cumpla con las especificaciones de los Libros Oficiales.
9.3.6 Del almacenamiento y sus condiciones
Los tanques o cisternas para almacenamiento de agua potable y de agua calidad farmacéutica deben cumplir con condiciones que aseguren su calidad. Para los mismos, debe haber procedimientos escritos para la limpieza, sanitización y control; debe registrarse la frecuencia, las acciones llevadas a cabo (rutinarias o correctivas) y los puntos de muestreo El almacenamiento del agua de calidad farmacéutica, no debe ser mayor de 24 horas o mantenerla en recirculación.
9.3.7 De los controles.
Deben realizarse y registrarse los controles fisicoquímicos y microbiológicos del agua potable y agua calidad farmacéutica, con la frecuencia necesaria.
9.4 Sistemas de aire
9.4.1 Generalidades.
Se debe mantener un sistema de tratamiento de aire que evite el riesgo de la contaminación física, química y biológica de los productos y las personas. Además, las condiciones de temperatura y humedad del aire, deben ajustarse a los requerimientos de los productos a elaborar y favorecer la comodidad de las personas. La ubicación del sistema debe facilitar la limpieza y mantenimiento.
9.4.2 De la contaminación. Los sistemas de aire para las áreas de producción deben evitar el riesgo de la contaminación cruzada entre los diferentes productos y procesos, para lo cual se debe incluir entre otras cosas filtros, pre filtros y todo equipo necesario para garantizar el grado de aire en un área de producción, (las rejillas de inyección y extracción, deben estar ubicadas de forma tal que el flujo del aire garantice el control de partículas según el área).
9.4.3 Del manejo del equipo. El sistema de aire debe contar con procedimientos escritos que abarquen las instrucciones y precauciones para su manejo.
9.4.4 De las operaciones de reparación y mantenimiento.
Debe existir un programa de mantenimiento preventivo documentado, que abarque los controles periódicos del sistema de aire que suministra a las diferentes áreas de producción. Debe establecerse la periodicidad para el cambio de filtros y pre filtros, con el fin de mantener su eficacia. Las operaciones de mantenimiento y reparación no deben presentar ningún riesgo para la calidad de los productos.
9.4.5 De los registros de mantenimiento del equipo. Debe mantenerse registros escritos del mantenimiento preventivo y correctivo de los equipos del sistema de aire.
9.4.6 De la destrucción de residuos y filtros del sistema de aire.
Deben existir procedimientos y registro para la destrucción de los residuos y filtros que se utilizaron en el sistema de inyección extracción de aire.
9.4.7 De los controles microbiológicos.
Deben realizarse controles microbiológicos de acuerdo al programa y procedimientos establecidos, para garantizar la calidad de aire de las áreas de producción y se deben mantener los registros respectivos.
10. MATERIALES Y PRODUCTOS
10.1 Generalidades
10.1.1 De los procedimientos.
Deben existir procedimientos escritos que describan en forma detallada la recepción, identificación, almacenamiento, manejo, muestreo, análisis y aprobación o rechazo de materiales y productos conforme a la especificación de cada uno de ellos.
10.1.2 Del manejo y almacenamiento.
Los materiales y productos, deben manejarse y almacenarse de tal manera que se evite cualquier contaminación o situación que pongan en riesgo la calidad de los productos.
10.1.3 De la ubicación.
Los recipientes o contenedores de materiales, deben mantenerse cerrados y ubicarse en tarimas o estantes, rotularse y separarse de las paredes, dejando el espacio suficiente para realizar su limpieza e inspección.
10.1.4 Del número de control.
Cada partida de materiales que ingrese a la empresa debe ser identificada con su correspondiente número de control, de acuerdo a la codificación establecida.
10.1.5 De los proveedores.
Los materiales deben proceder solamente de proveedores aprobados, mencionados en la especificación correspondiente y cuando sea posible, directamente del productor, y las especificaciones establecidas por el laboratorio fabricante para los materiales se deben discutir con los proveedores.
10.1.6 De la integridad de los recipientes.
En cada entrega de material se comprobará la integridad de los recipientes y sus cierres, así como la correspondencia entre la nota de entrega y las etiquetas del proveedor.
10.1.7 De la cuarentena.
Cada lote de los materiales y productos debe permanecer en cuarentena mientras no sea muestreado, examinado y analizado por Control de Calidad, quien debe emitir su aprobación o rechazo.
10.1.8 Del muestreo.
Se deben tomar muestras, estadísticamente representativas, de cada ingreso de materiales. Las muestras de materia prima deben ser retenidas, por lo menos durante un año, después de la fecha de expiración del último lote del producto fabricado, que contenga el ingrediente.
10.1.9 Del muestreo de diferentes lotes.
Si una entrega de material está compuesta por diferentes lotes, cada lote debe considerarse por separado para efectos de muestreo, análisis y aprobación.
10.1.10 De la identificación de materiales
Cada lote del material estará debidamente identificado con una etiqueta que incluya como mínimo:
a. Nombre y código del material.
b. Número de ingreso asignado por el establecimiento receptor para cada lote en cada entrega recibida.
c. Situación del material (cuarentena, aprobado, rechazado)
d. Nombre del proveedor.
e. Fecha de expiración.
f. Número de análisis.
10.2 Materias primas
10.2.1 De la integridad de los recipientes.
Cada lote de materia prima debe ser inspeccionado visualmente, para verificar su estado físico, al momento de recibirla. El sistema de cierre debe garantizar la integridad, su inviolabilidad e identidad.
10.2.2 De la identificación y estado.
Cada lote de materia prima estará debidamente identificado con una etiqueta que incluya como mínimo:
a. Nombre de la materia prima
b. Código interno
c. Nombre del fabricante. d. Nombre del proveedor.
e. Cantidad del material ingresado.
f. Código o número de lote del fabricante.
g. Fecha de expiración. h. Condiciones de almacenamiento.
i. Advertencias y precauciones.
j. Fecha de análisis
k. Fecha de re-análisis, siempre y cuando no haya expirado.
l. Estado o situación (cuarentena, muestreado, aprobado o rechazado).
m. Observaciones.
En caso de que los sistemas de almacenamiento hayan sido totalmente computarizados, no es necesario que toda la información mencionada figure en la etiqueta en forma impresa.
10.2.3 Del trasvasado de la materia prima.
Si una materia prima es removida del envase original y trasvasado a otro envase, el nuevo recipiente debe cumplir con los requisitos de identidad establecidos en el anterior. Se debe dejar registro de la sustancia contenida anteriormente, es recomendable utilizar un recipiente que haya contenido la misma materia prima o sustancia.
10.2.4 De la autorización para su uso.
Cada materia prima debe ser muestreada, examinada y analizada de acuerdo a procedimientos escritos. Si cumple con las especificaciones será aprobada y autorizada para su uso. De no ser así, la materia prima será rechazada.
10.2.5 Casos especiales.
Cuando la materia prima ha estado expuesta al aire, temperatura extrema, humedad o cualquier otra condición que pudiera afectarla negativamente, debe separarse e identificarse de inmediato según los procedimientos de manejo de materias primas. Control de calidad debe proceder a la aprobación o rechazo, de acuerdo con los resultados obtenidos del análisis.
10.2.6 De la utilización.
Sólo podrán utilizarse las materias primas aprobadas por el departamento de control de calidad y que no hayan expirado.
10.2.7 Del dispensado.
Las materias primas deben ser fraccionadas por personal designado a tal fin, de acuerdo a un procedimiento escrito que garantice que se pesen o midan de forma precisa y exacta, en recipientes limpios e identificados.
10.2.8 De la identificación de la materia prima dispensada.
Cada recipiente conteniendo materia prima dispensada de identificarse con una etiqueta con la siguiente información como mínimo:
a. Nombre de la materia prima.
b. Código o número de lote o número de ingreso.
c. Nombre del producto a fabricar.
d. Código de lote del producto a fabricar.
e. Contenido neto (SI sistema internacional de unidades de medida).
f. Fecha de dispensado.
g. Nombre y firma de la persona que dispenso.
h. Nombre y firma de la persona que reviso.
10.2.9 De la verificación de pesado o medida.
La materia prima pesada o medida, debe ser verificada por otra persona autorizada y debe quedar registrada.
10.2.10 De las materias primas dispensadas. Las materias primas para ser utilizadas en cada lote de producción deben mantenerse agrupadas e identificadas de forma visible, para evitar riesgos de confusión y contaminación.
10.3 Materiales de acondicionamiento
10.3.1 De los envases y cierres primarios.
Los envases y cierres primarios deben ser diseñados con un material que no sea reactivo, aditivo o absorbente y así evitar alteraciones en la seguridad, identidad, potencia o pureza del producto en todo momento. Los requerimientos de los envases y cierres primarios deben estar sustentados en los estudios de formulación, pruebas de estabilidad y aprobación de proveedores
10.3.2 De la manipulación.
Los envases, cierres y medidas dosificadoras, deben estar limpios y manipularse de acuerdo a procedimiento escrito.
10.3.3 De la dispensación.
Todos los materiales de acondicionamiento deben ser examinados respecto a su cantidad, identidad, y conformidad con las respectivas instrucciones de la orden de envasado, antes de ser enviados al área.
10.3.4 De los materiales impresos.
Estos materiales se conservarán bajo llave y acceso restringido de forma que evite el ingreso de personas no autorizadas al mismo. Las etiquetas y material impreso deben manipularse de tal forma que se evite cualquier confusión
10.4 Productos intermedio y a granel
10.4.1 De la manipulación.
Todos los productos intermedios y a granel, deben manipularse y almacenarse de tal manera que se evite cualquier contaminación o que ponga en riesgo la calidad de los productos.
10.5 Productos terminados
10.5.1 De la cuarentena.
Deben mantenerse en cuarentena hasta su aprobación final en las condiciones establecidas por el laboratorio fabricante.
10.5.2 Del producto terminado aprobado.
Los productos terminados pueden ser comercializados solamente después de su aprobación.
10.6 Materiales y productos rechazados
10.6.1 De la manipulación.
Deben existir procedimientos escritos para la manipulación de materiales, productos intermedios a granel y productos terminados que han sido rechazados. Los cuales deben identificarse, mediante el uso de una etiqueta roja justificando la causa del rechazo.
10.6.2 Del destino de los materiales.
Los materiales rechazados serán devueltos a los proveedores o destruidos, de acuerdo al procedimiento establecido por el laboratorio fabricante y debe cumplir con la normativa ambiental existente en cada estado parte.
10.6.3 De la disposición del material obsoleto o desactualizado.
Debe ser identificado, manipulado y destruido según procedimiento, dejando registros.
10.7 Productos devueltos
10.7.1 Del procedimiento. Debe existir un procedimiento escrito para la devolución de producto.
10.7.2 Del almacenamiento.
Los productos devueltos deben ser identificados y almacenados en un área separada y de acceso restringido, de acuerdo a un procedimiento escrito.
10.7.3 Del manejo.
Los productos devueltos serán manejados de acuerdo a un procedimiento escrito establecido para este fin. En dicho procedimiento se definirá quien es el responsable de esta acción conjuntamente con Garantía de Calidad o Control de Calidad.
10.7.4 De las condiciones extremas.
Los productos farmacéuticos devueltos por envejecimiento o que hayan sido sometidos a condiciones inadecuadas de manejo o almacenamiento, incluyendo temperatura extrema, humedad, humo, gases, presión, radiación o cualquiera otra situación perjudicial, deben ser destruidos, según procedimiento escrito.
10.7.5 De los registros.
Se deben mantener registros de los productos devueltos con el nombre, forma farmacéutica, número de lote, motivo de la devolución, cantidad devuelta y fecha de la devolución.
10.7.6 De la investigación.
Una vez determinada la causa de la devolución, se debe investigar si esta pudo afectar cualquier otro lote dejando registro de las acciones correctivas y el seguimiento de la devolución.
11. DOCUMENTACIÓN
11.1 Generalidades
11.1.1 De las generalidades.
La documentación es parte esencial del Sistema de Garantía de Calidad, debe considerarse en todos los aspectos de las Buenas Prácticas de Manufactura. La documentación escrita claramente evita errores propios de la comunicación oral y permite seguir la historia de los lotes. Las especificaciones, fórmulas, métodos e instrucciones de fabricación, procedimientos y registros deben estar en forma impresa, debidamente revisadas y aprobadas. La legibilidad de los documentos es de importancia primordial.
11.1.2 Del diseño.
Todos los documentos deben de ser diseñados, revisados, y distribuidos cuidadosamente de acuerdo al procedimiento.
11.1.3 De la aprobación. Los documentos deben ser aprobados, firmados y fechados por las personas autorizadas. Ningún documento debe modificarse sin autorización.
11.1.4 De las características de los documentos.
Los documentos deben:
a. Redactarse en forma clara, ordenada y libre de expresiones ambiguas permitiendo su fácil comprensión.
b. Ser fácilmente verificables.
c. Revisarse periódicamente y mantenerse actualizados.
d. Ser reproducidos en forma clara, legible e indeleble.
11.1.5 Del registro de datos en documentos
La introducción de datos, puede realizarse escribiendo a mano con letra clara, legible y con tinta indeleble. Debe dejarse espacio suficiente para permitir la realización del registro de datos.
11.1.6 De otras formas de registro de los datos
Los documentos y los datos pueden estar registrados en forma impresa, por medios electrónicos o por medio de otro sistema. En el caso de almacenar la información de forma electrónica deben crearse controles especiales. Si la documentación es realizada por el método de procesamiento electrónico de datos, solo las personas autorizadas deben accesar o modificar los datos en la computadora y debe existir un registro de los cambios y las eliminaciones; el acceso debe de estar restringido por contraseñas u otros medios y la entrada de los datos críticos debe ser verificada.
11.1.7 De las correcciones de datos en documentos
Cualquier corrección realizada en un dato escrito de un documento debe firmarse y fecharse; la corrección no debe impedir la lectura del dato inicial. Cuando sea necesario, habrá que indicar la causa de la corrección.
11.1.8 De la trazabilidad.
Debe mantenerse registro de todas las acciones efectuadas o completadas de tal forma que haya trazabilidad de las actividades significativas relativas a la fabricación de productos farmacéuticos. Todos los registros incluyendo lo referente a los procedimientos de operación, deben mantenerse por un año, como mínimo, después de la fecha de expiración del producto terminado.
11.1.9 Del listado. Se debe establecer un listado maestro de documentos fácilmente disponible, que identifique el estado de los mismos.
11.1.10 De la ubicación.
Se debe contar con ediciones actualizadas de los documentos, en todos los sitios donde se efectúen operaciones esenciales para el desempeño del proceso.
11.1.11 De los documentos obsoletos.
Se debe evitar la utilización de documentos invalidados u obsoletos. Éstos se deben retirar de todos los puntos de uso. El original del documento obsoleto se debe mantener en un archivo histórico identificado.
11.2 Documentos exigidos.
11.2.1 De las especificaciones.
Debe disponerse de especificaciones autorizadas y fechadas por control de calidad para la materia prima, material de envase, empaque, productos intermedios o granel y producto terminado.
11.2.2 Del contenido de las especificaciones de materiales y producto terminado.
Las especificaciones de la materia prima, material de envase, empaque, productos intermedios o granel y producto terminado, debe incluir:
a. Nombre del material o producto, (Denominación Común Internacional, cuando corresponda).
b. Código de referencia interna.
c. Referencia, si la hubiere, de los libros oficiales.
d. Fórmula química (cuando aplique).
e. Requisitos cuali y cuantitativos con límites de aceptación (cuando aplique).
f. Las técnicas analíticas o procedimiento. g. Procedimiento de muestreo.
h. Muestra del material impreso (cuando aplique).
i. Cantidad requerida para la muestra de retención.
j. Condiciones de almacenamiento y precauciones.
k. Proveedores aprobados y marcas comerciales (cuando aplique)
l. Descripción de la forma farmacéutica y detalles de empaque (cuando aplique).
m. Vida en anaquel (cuando aplique).
11.2.3 De la actualización.
Debe haber una revisión periódica de las especificaciones analíticas, usando de referencia los libros oficiales.
11.2.4 De la fórmula maestra.
Se debe contar con una formula maestra para cada producto y tamaño del lote a fabricar.
11.2.5 Del contenido de la fórmula maestra.
La Fórmula Maestra debe incluir:
a) El nombre y código del producto correspondiente a su especificación.
b) Una descripción de la forma farmacéutica, concentración del principio activo principio activo y tamaño del lote.
c) Fórmula cualicuantitativa expresada en unidades del sistema internacional, de las materias primas a emplearse, (debe hacer mención de cualquier sustancia que pueda desaparecer durante el proceso), usando el nombre y código que es exclusivo para cada material.
d) Una lista de material de empaque primario y secundario a emplearse, indicando la cantidad de cada uno, usando el nombre y código que es exclusivo para cada material.
e) Una indicación del rendimiento esperado con los límites de aceptabilidad y de los rendimientos intermedios pertinentes, en los casos que corresponda.
f) Indicación del área y de los principales equipos a ser empleados.
g) Instrucciones detalladas de los pasos a seguir en el proceso de producción.
h) Instrucciones referentes a los controles durante el proceso, con sus límites.
i) Cuando fuere necesario, instrucciones para el almacenamiento de los productos, incluyendo el contenedor, el etiquetado y cualesquiera otras condiciones de almacenamiento.
j) Precauciones especiales que deben tomarse en cuenta.
k) Nombre, firma y fecha de las personas responsables de aprobar la fórmula maestra.
11.2.6 De la relación entre Fórmula Maestra y el Protocolo de Registro Sanitario.
Las Fórmulas Maestras de todos los productos fabricados, deben coincidir con las fórmulas presentadas en la documentación para obtención del registro sanitario.
11.2.7 De la emisión de la orden de producción.
La orden de producción correspondiente a un lote debe ser emitida por el departamento de producción o cualquier otra instancia superior, según la organización de la empresa conteniendo la información necesaria para la producción, la cual debe ser una copia exacta del registro de la fórmula maestra (que al asignarle un código de lote se convierte en orden de producción). Debe estar autorizada por los responsables de los departamentos involucrados.
11.2.8 Del contenido de la orden y del registro de producción.
Además de lo indicado en la formula maestra debe contener lo siguiente:
a. Código o número de lote.
b. Fecha de inicio y finalización de la producción.
c. Fecha de expiración del producto.
d. Firma de las personas que autorizan la orden de producción.
e. Número de lote de la materia prima.
f. Firma de la persona que despacha, recibe y verifica los insumos.
g. Firma de las personas que intervienen y supervisan los procesos.
h. Resultados de los análisis del producto en proceso.
i. Hojas para el registro de controles durante el proceso y espacio para anotar observaciones.
j. Declaración del rendimiento teórico previsto con los límites de aceptación, y de rendimientos intermedios significativos, en su caso.
k. Indicaciones de las precauciones necesarias para el almacenamiento del producto a granel si fuera necesario.
l. Instrucciones para la toma de muestras en las etapas que sean necesaria.
11.2.9 Del contenido de la orden de envasado y empaque
Además de lo indicado en la formula maestra, la orden de envasado y empaque debe incluir lo siguiente:
a. Código o número de lote.
b. Cantidad del producto a envasar o empacar.
c. Fecha de inicio y finalización de las operaciones de acondicionamiento.
d. Fecha de expiración del producto.
e. Firma de las personas que autorizan la orden de envase y empaque.
f. Número de lote de cada material de envase y empaque utilizado.
g. Firma de la persona que despacha, recibe y verifica los insumos.
h. Firma de las personas que intervienen y supervisan los procesos.
i. Hojas para el registro de controles durante el proceso y espacio para anotar observaciones.
j. Muestras del material de acondicionamiento impreso que se haya utilizado, incluyendo muestras con el número de lote, fecha de expiración y cualquier impresión suplementaria.
k. Cantidades de los materiales impresos de acondicionamiento que han sido devueltos al almacén o destruidos y las cantidades de producto obtenido, con el fin de obtener el balance adecuado, 1. Número de registro sanitario.
11.3 Procedimientos y registros
11.3.1 De los procedimientos.
Se debe contar con procedimientos escritos para el control de la producción y demás actividades relacionadas. Estos documentos deben ser registrados durante la ejecución de las funciones respectivas, debiendo quedar escritos y firmados de conformidad con el registro de firmas, inmediatamente después de su realización. Cualquier desviación de los procedimientos que pueda afectar la calidad del producto por un evento atípico, debe quedar registrada y justificada.
11.3.2 De los registros de los lotes.
Cada lote de producto debe generar registros de producción y control, para garantizar el cumplimiento de los procedimientos escritos y aprobados.
11.3.3 De la revisión de los registros de los lotes
Control de calidad o garantía de calidad debe revisar y aprobar todos los registros de producción y control de cada lote terminado, para verificar el cumplimiento de los procedimientos escritos y aprobados. Cualquier desviación no justificada debe ser ampliamente investigada, la cual debe extenderse a otros lotes afectados y a otros productos que puedan estar asociados con la discrepancia encontrada.
11.3.4 Del archivo y conservación
Debe existir un procedimiento escrito para el archivo y conservación de la documentación de un lote cerrado de producción.
11.3.5 De los procedimientos.
Debe disponerse de procedimientos escritos y registros correspondientes de las actividades realizadas sobre:
a. Mantenimiento, limpieza y sanitización de edificios e instalaciones.
b. Uso, mantenimiento, limpieza y sanitización de equipos y utensilios.
c. Sanitización y mantenimiento de tuberías y de las tomas de fluidos.
d. Calibración de equipo.
e. Asignación de número de lote
f. Capacitación del personal,
g. Uso y lavado de uniformes,
h. Control de las condiciones ambientales,
i. Prevención y exterminio de plagas (contemplando la no contaminación de equipos, materias primas, material de acondicionamiento y producto terminado, con insecticidas, agentes de fumigación y materiales de saneamiento).
j. Recolección, clasificación y manejo de basuras y desechos.
k. Muestreo.
l. Validaciones,
m. Cualquier otro que sea necesario.
12. PRODUCCIÓN
12.1 Generalidades
12.1.1 De las generalidades.
De conformidad con las autorizaciones de fabricación, en las operaciones de producción se debe cumplir procedimientos claramente definidos con el objeto de obtener productos que reúnan las condiciones de calidad exigidas.
12.1.2 De las operaciones.
Todas las operaciones de manejo de materiales y productos, tales como cuarentena, muestreo, almacenamiento, etiquetado, despacho, elaboración, envasado y distribución, deben efectuarse de conformidad con procedimientos o instrucciones escritas y debidamente registradas.
12.1.3 De las desviaciones.
Debe evitarse cualquier desviación de las instrucciones o procedimientos, cuando haya que efectuar alguna desviación, está debe ser aprobada por escrito por la persona asignada con participación del Departamento de Control de Calidad.
12.1.4 Del reproceso.
Solo en casos excepcionales habrá de reprocesarse los productos rechazados. Será permitido solamente si no se ve afectada la calidad del producto, si reúnen todas las especificaciones, y si se efectúa de conformidad con un proceso bien definido y autorizado, una vez realizada la evaluación de los riesgos existentes. Se debe registrar el reprocesado y asignarse un nuevo código o número al lote reprocesado. El producto no podrá ser reprocesado una vez que haya salido del laboratorio fabricante.
12. 1.5 De los registros.
Deben mantenerse registros de los controles efectuados durante el proceso, los cuales formaran parte de los registros de los lotes
12.1.6 Del uso del área.
No deben llevarse a cabo operaciones simultáneas con diferentes productos en la misma área, a excepción del área de empaque secundario con sus líneas definidas y separadas.
12.1.7 De la identificación.
Durante todo el proceso, todos los materiales, producto a granel, equipos y áreas utilizadas, deberán identificarse como mínimo con: nombre del producto que se esté elaborando, código o número de lote y fase del proceso.
12.1.8 Del muestreo.
La toma de la muestra de los productos intermedios y de los productos terminados, debe basarse en criterios estadísticos relacionados con la variabilidad del proceso, los niveles de confiabilidad y el grado de precisión que se requiere. Realizándose en la misma área de producción.
12.1.9 Del uso exclusivo de las áreas y equipo de producción.
No se debe utilizar las áreas y el equipo destinado a la producción de medicamentos para producir otro tipo de productos.
12.2 Prevención de la contaminación cruzada y microbiana en la producción
12.2.1 De la contaminación.
Se debe evitar la contaminación en todas las fases de producción, los productos y materiales deberán protegerse de la contaminación microbiana o de otro tipo de contaminación que incida en la calidad.
12.2.2 De la contaminación cruzada.
La contaminación cruzada debe evitarse mediante las medidas técnicas o procedimientos, tales como:
a. Existencia de esclusas. (cuando aplique)
b. Áreas con diferenciales de presión
c. Sistemas de inyección-extracción que garanticen la calidad de aire.
d. Uso de ropa protectora dentro de las áreas en las que se elaboren productos con riesgo especial de contaminación cruzada.
e. Uso de procedimientos de limpieza y sanitización.
f. Pruebas para detectar residuos (trazas), en los productos altamente activos.
g. Utilización de etiquetas con la situación del estado de limpieza del equipo y áreas
12.2.3 De la verificación de las medidas adoptadas.
Debe verificarse periódicamente la eficacia de las medidas destinadas a prevenir la contaminación cruzada. Dicha verificación se debe hacer de conformidad con procedimientos de operación.
12.2.4 De la contaminación microbiológica.
Para productos no estériles, se deben establecer y cumplir procedimientos escritos y validados, para evitar la contaminación con microorganismos patógenos y mantener los recuentos microbianos dentro de especificaciones.
12.3 Controles en proceso
12.3.1 Del despeje de línea.
Antes de iniciar las operaciones de producción se debe verificar, registrar y documentar todas las acciones requeridas para asegurar que el equipo y el área estén limpios para su utilización y se encuentre libre de productos, documentos y materiales no necesarios para las operaciones previstas.
12.3.2 De los controles durante el proceso.
Los controles durante el proceso deben realizarse dentro de las áreas de producción siempre que no pongan en riesgo la producción.
12.3.3 De los controles en línea.
El control del producto en línea, durante el envasado y empaque debe incluir al menos los siguientes:
a. Aspecto general de los envases;
b. La cantidad de material de acondicionamiento este completo y correcto.
c. El lote y la fecha de expiración están correctos y legibles.
d. Funcionamiento adecuado de los controles de línea.
e. Verificar la integridad de los sellos.
12.3.4 De los controles microbiológicos.
Deben realizarse controles microbiológicos de superficie de acuerdo a un programa y procedimiento establecido. Se deben mantener los registros apropiados.
12.3.5 De los controles ambientales.
Se deben llevar a cabo y registrarse los controles ambientales.
12.3.6 Del material impreso.
El material impreso para la rotulación de cada lote, debe ser cuidadosamente inspeccionado para verificar y documentar, que su identidad corresponde con la rotulación especificada en el registro de producción.
12.3.7 Del tiempo de etiquetado.
El etiquetado debe efectuarse después del envasado y cierre, si se demora se deben tomar medidas para asegurar que no haya confusión o errores en el etiquetado.
12.3.8 De las muestras tomadas.
Las muestras tomadas de la línea de envasado y empaque no deben volver a la misma.
12.3.9 De las desviaciones de rendimiento.
Cualquier desviación significativa del rendimiento esperado debe ser registrada e investigada.
12.3.10 De la conciliación.
Se deben establecer procedimientos escritos, para conciliar las cantidades de etiquetas entregadas, usadas, devueltas en buen estado y destruidas. Se debe realizar una evaluación de las diferencias encontradas e investigar sus causas. Los resultados, conclusiones y acciones correctivas deben registrarse.
13 GARANTÍA DE CALIDAD.
13.1 Generalidades
13.1.1 De las generalidades
Garantía de calidad es responsabilidad de la dirección de la empresa y exige la participación y el compromiso del personal de los diferentes departamentos y a todos los niveles dentro de la empresa. Para asegurar la calidad es necesaria la existencia de una política de calidad definida y documentada en un sistema de garantía de calidad.
13.1.2 Garantía de calidad.
El sistema de Garantía de Calidad debe asegurar que:
a. Los medicamentos se diseñan y desarrollan de forma que se tenga en cuenta lo requerido por las Buenas Prácticas de Manufactura y las Buenas Prácticas de Laboratorio, disponiéndose de los protocolos y registros correspondientes.
b. Las operaciones de producción y control deben estar claramente especificadas de acuerdo con las Buenas Prácticas de Manufactura.
c. Las responsabilidades del personal directivo estén claramente especificadas y divulgadas.
d. Se tengan requisitos establecidos para el abastecimiento y utilización de la materia prima, materiales de envase y empaque y en la preparación de los productos.
e. Se realiza una evaluación y aprobación de los diferentes proveedores.
f. Se realizan todos los controles necesarios de los productos intermedios y cualquier otro tipo de controles durante el proceso.
g. El producto terminado se ha elaborado y controlado de forma correcta, según procedimientos definidos.
h. Exista un procedimiento para la recopilación de la documentación del producto que se ha elaborado.
i. Los medicamentos no se venden o suministran antes de que una persona calificada haya aprobado que cada lote de producción se ha fabricado y controlado de acuerdo a los requisitos de la autorización de comercialización.
j. Se tomen medidas adecuadas para asegurar, que los medicamentos sean almacenados y distribuidos de manera que la calidad se mantenga durante todo el período de vida útil.
k. Existe un procedimiento de auto inspección y auditoría de la calidad que evalúa periódicamente la efectividad y aplicabilidad del sistema de Garantía de Calidad.
l. Existan procedimientos, programas y registros de los Estudios de Estabilidad de los productos, los cuales garanticen las condiciones apropiadas de manejo, almacenamiento y fecha de expiración.
m. Exista un plan maestro de validación y su cumplimiento
14. CONTROL DE CALIDAD.
14.1 Generalidades.
14.1.1 De las generalidades.
Control de calidad como parte de Buenas Prácticas de Manufactura, debe de contar con la documentación necesaria que asegure que el suministro de materiales y la comercialización de productos, se realice hasta que su calidad haya sido aprobada.
14.1.2 De control de calidad.
El laboratorio fabricante debe contar con una unidad de Control de Calidad la cual no debe limitarse a operaciones de laboratorio, sino que debe intervenir en todas las decisiones que afecte Ja calidad del producto.
14.1.3 De la independencia.
La unidad de Control de Calidad debe ser independiente de producción y estar bajo la responsabilidad de un profesional calificado. Contará con los recursos adecuados que garanticen que todas las decisiones se realicen de forma confiable.
14.1.4 De las obligaciones.
La unidad de control de calidad tendrá, entre otras obligaciones las siguientes: establecer, validar, verificar y aplicar todos los procedimientos de control de calidad, conservar las muestras de referencia de materiales y productos, garantizar el etiquetado correcto de los envases, de materiales y productos, realizar la estabilidad de los productos, participar en la investigación de reclamos relativos a la calidad del producto y cualquier otra actividad pertinente a las operaciones de control de calidad, estas se realizarán de acuerdo a procedimientos escritos y deben quedar registradas.
14.1.5 De la aprobación de los productos terminados.
La aprobación de cada lote de productos terminados, debe realizarlo la persona responsable de la misma, después de evaluar debidamente que, dicho lote está conforme con las especificaciones establecidas, incluyendo las condiciones de producción, análisis en proceso y la documentación para su aprobación final.
14.1.6 De las desviaciones.
Cualquier desviación de los parámetros escritos debe ser investigada y documentada, dando seguimiento a las acciones correctivas
14.1.7 Del acceso del personal de control de calidad.
El personal de control de calidad debe tener acceso a las áreas de producción con fines de muestreo, inspección, investigación y otros trabajos relacionados con las Buenas Prácticas de Manufactura.
14.1.8 De los equipos de control de calidad.
La unidad de control de calidad debe contar con el equipo necesario para realizar los análisis requeridos. En caso de no contar con equipo requerido para efectuar análisis específicos, debe contratar los servicios analíticos de un laboratorio de Control de Calidad debidamente autorizado.
14.1.9 Del mantenimiento y calibración del equipo.
El equipo de la unidad de control de calidad debe tener programas y registros escritos de mantenimiento, verificación y calibración, para asegurar su funcionamiento correcto y la validez de los resultados.
14.2 Documentación
14.2.1 De los documentos de control de calidad.
La unidad de control de calidad debe tener como mínimo a su disposición lo siguiente:
a. Especificaciones de toda materia prima y material de acondicionamiento.
b. Procedimiento para manejo de muestra de retención.
c. Metodología analítica para el análisis de materia prima y producto terminado, con su referencia.
d. Procedimientos de control y resultados de las pruebas (incluyendo los documentos de trabajo utilizados en el análisis y registros de laboratorio).
e. Informes/ certificados analíticos.
f. Registro de las condiciones ambientales, cuando aplique.
g. Procedimientos y registros de validación de los métodos de ensayo.
h. Procedimientos y registros para la calibración de instrumentos y equipos. i. Procedimientos y registros del mantenimiento del equipo.
j. Procedimiento de selección y calificación de proveedores.
k. Procedimiento y programa de sanitización de áreas.
l. Procedimiento para el uso de instrumental.
m. Procedimiento para la aprobación y rechazo de materiales y producto terminado.
n. Procedimiento para el mantenimiento de instalaciones de control de calidad.
ñ. Procedimiento para el manejo y desecho de solventes.
o. Procedimiento para la recepción, identificación, preparación, manejo y Almacenamiento de reactivos y estándares.
p. Procedimiento para el lavado de cristalería.
q. Cualquier otro procedimiento que sea necesario en la unidad de Control de Calidad.
14.2.2 Del archivo de control de calidad.
Cualquier documentación de control de calidad relativa a un Jote debe conservarse según la legislación de cada país.
14.3 Muestreo.
14.3.1 Del procedimiento de muestreo.
El procedimiento debe contener como mínimo lo siguiente:
a. El método de muestreo
b. El equipo que debe utilizarse
c. La cantidad de muestra que debe recolectarse
d. Instrucciones para la eventual subdivisión de la muestra
e. Tipo y condiciones del envase que debe utilizarse para la muestra
f. Identificación de los recipientes muestreados.
g. Precauciones especiales que deben observarse, especialmente en relación con el muestreo de material estéril o de uso delicado.
h. Condiciones de almacenamiento
i. Instrucciones de limpieza y almacenamiento del equipo de muestreo
14.3.2 De la representatividad de las muestras.
La cantidad de muestra debe ser estadísticamente representativa del lote de materiales, productos intermedios y productos terminados.
14.3.3 De la identificación de las muestras.
Las etiquetas deben indicar:
a. Nombre del material o producto.
b. Cantidad.
c. Número de lote.
d. Fecha de muestreo.
e. Recipientes de los que se han tomado las muestras.
f. Nombre y firma de la persona que realiza el muestreo.
14.3.4 De las muestras de referencia.
Deben conservarse muestras de referencias de cada lote de ingredientes activos y producto terminado hasta un año después de la fecha de expiración, en cantidad suficiente para permitir un análisis completo. Los productos terminados, se conservarán normalmente en su empaque final y se mantendrán en las condiciones de almacenamiento según especificación del producto.
14.4 Metodología analítica
14.4. 1 De los métodos
Los métodos analíticos deben estar escritos, aprobados y validados.
14.4.2 Del registro de datos de los análisis.
Los resultados de los análisis realizados quedarán registrados en los correspondientes protocolos que incluirán, como mínimo los siguientes datos:
a. Nombre del material o producto.
b. Forma farmacéutica (cuando aplique).
c. Presentación farmacéutica (cuando aplique).
d. Número de lote. e. Nombre del fabricante y proveedor, cuando se declare.
f. Referencias de las especificaciones y procedimientos analíticos pertinentes.
g. Resultados de los análisis, con observaciones, cálculos, gráficas, cromatogramas y referencias.
h. Fechas de los análisis.
i. Firma registrada de las personas que realicen los análisis.
j. Firma registrada de las personas que verifiquen los análisis y los cálculos.
k. Registro de aprobación o rechazo (u otra decisión sobre la consideración del producto), fecha y firma del responsable designado.
14.4.3 De los controles.
Todos los controles durante el proceso de producción, deben ser llevados a cabo por personal asignado en dicho proceso, de acuerdo a los métodos aprobados por control de calidad y sus resultados quedarán registrados.
14.4.4 De los reactivos químicos, medios de cultivo, patrones y cepas de referencia
Los reactivos químicos, medios de cultivos, patrones y cepas de referencia deben ser preparados, identificados, trazables, conservados y utilizados de acuerdo con instrucciones definidas y escritas, manteniendo un control sobre las fechas de expiración.
14.4.5 De la rotulación de reactivos. Los reactivos de laboratorio se rotularán con la fecha de preparación, fecha de vencimiento y la firma de la persona que los preparo, detallando las condiciones específicas de almacenamiento. Además, en el caso de las soluciones volumétricas, se indicará la última fecha de valoración y concentración.
14.5 Estabilidad
14.5.1 De las generalidades.
La unidad de control de calidad debe realizar evaluación de los productos farmacéuticos terminados, con el fin de garantizar que el producto cumpla con las especificaciones de calidad durante su vida útil. La estabilidad debe determinarse antes de la comercialización y también después de cualquier modificación significativa en la fabricación de los productos, aplicando los métodos de estabilidad acelerada, estabilidad en estante o de largo plazo según la normativa vigente.
14.5.2 Del programa y protocolo.
La unidad de control de calidad debe de contar con un programa permanente y protocolos, para la determinación de la estabilidad de los productos, que incluya:
a. Descripción completa del producto.
b. Parámetros y métodos de pruebas.
c. Cantidad suficiente del producto
d. Cronograma de pruebas.
e. Condiciones especiales de almacenamiento.
f. Resumen de todos los datos obtenidos (informe, cálculos, conclusiones).
14.5.3 De la vida útil
Se deben establecer fechas de caducidad y condiciones de almacenamiento basado en el estudio de estabilidad.
15 PRODUCCIÓN Y ANÁLISIS POR CONTRATO.
15.1 Generalidades.
15.1.1 De las generalidades
La producción y análisis por contrato debe definirse, aprobarse y controlarse correctamente, Existiendo un contrato por escrito entre el contratante y el contratista (contratado) en el que se establezcan claramente las obligaciones de cada parte.
15.1.2 De contratos a terceros
La producción y el análisis de productos por terceros deben ser definidos, de mutuo consentimiento por medio de un contrato, mediante instrumento público debidamente autorizado.
15.1.3 De las obligaciones del contrato.
El contrato debe estipular claramente las obligaciones de cada una de las partes, con relación a la fabricación, manejo, almacenamiento, control y liberación del producto.
15.1.4 Del responsable.
En el contrato se debe establecer claramente la persona responsable de autorizar la liberación de cada lote para su comercialización y de emitir el certificado de análisis.
15.1.5 Del contenido del contrato.
El contrato a terceros deberá contemplar como mínimo los siguientes aspectos:
a. Debe ser redactado por personas competentes y autorizadas.
b. Aceptación de los términos del contrato por las partes.
c. Aceptación del cumplimiento del Reglamento Centroamericano en Buenas Prácticas de Manufactura.
d. Abarcar la producción y el análisis o cualquier otra gestión técnica relacionada con estos.
e. Debe describir el manejo de materias primas, material de acondicionamiento, gráneles y producto terminado, en caso sean rechazados.
f. Permitir el ingreso del contratante a las instalaciones del contratista (contratado), para auditorias.
g. Permitir el ingreso del contratista (contratado) a las instalaciones del contratante.
h. Listar cada uno de los productos o servicios de análisis objeto del contrato.
15.1.6 De la inspección a las instalaciones del contratista.
En caso de análisis por contrato, el contratista (contratado) debe aceptar que puede ser inspeccionado por la Autoridad Reguladora.
15.2 Del contratante.
15.2.1 De las responsabilidades del contratante.
El contratante debe asegurarse que el contratista (contratado):
a. Cumpla con los requisitos legales, para su funcionamiento.
b. Cumpla con las buenas prácticas de manufactura y de laboratorio, con instalaciones, equipo, conocimientos y experiencia para llevar a cabo satisfactoriamente el trabajo contratado.
c. Posea certificado vigente de buenas prácticas de manufactura.
d. Entregue los productos elaborados cumpliendo con las especificaciones correspondientes y que han sido aprobados por una persona calificada. e. Entregue los certificados de análisis con su documentación de soporte, cuando aplique según contrato.
15.3 Del contratista.
15.3.1 De las responsabilidades del contratista (contratado).
El contratista (contratado) debe asegurarse que el contratante:
a. Cumpla con los requisitos legales para su funcionamiento.
b. Trámite y obtenga el registro sanitario del producto a fabricar.
c. Proporcione toda la información necesaria para que las operaciones se realicen de acuerdo al registro sanitario y otros requisitos legales.
15.3.2 De las limitaciones
El contratista no podrá ceder a un tercero en todo o en parte el trabajo que se le ha asignado por contrato. Además, debe abstenerse de llevar a cabo cualquier actividad que pueda afectar la calidad del producto fabricado o analizado.
16. VALIDACIÓN
16.1 Generalidades
16.1.1 De las generalidades.
Los estudios de validación constituyen una parte esencial de las buenas prácticas de manufactura y deben efectuarse de acuerdo a un plan maestro y su programa quedando registrados los resultados y conclusiones.
16.1.2 De la organización.
Debe existir un comité multidisciplinario responsable de coordinar e implementar el plan maestro y todas las actividades de validación.
16.2 De la conformación de equipos
Deben existir equipos conformados por expertos en los diferentes aspectos a validar.
16.3 De los protocolos e informes.
Deben existir protocolos de validación que describan el procedimiento a seguir para la realización de la validación y un informe final o dictamen que resuma los resultados y conclusiones obtenidas, los mismos deben estar debidamente autorizados y archivados.
16.4 De la calificación y validación.
Se deben realizar y documentar las calificaciones y validaciones de:
a. Equipos de producción y control de calidad.
b. Métodos analíticos.
c. Procesos de producción de no estériles.
d. Procesos de producción de estériles.
e. Procedimientos de limpieza.
f. Sistemas de agua.
g. Sistemas de aire.
h. Sistemas de vapor (calderas, marmitas y otros)
i. Instalaciones
j. Sistemas informáticos, cuando aplique.
16.5 De nueva fórmula.
Cuando se adopte una nueva fórmula o método de preparación, se deben tomar medidas para demostrar que las modificaciones realizadas dan como resultado un producto que consistentemente posee la calidad exigida.
16.6 De la validación de modificaciones.
Debe validarse las modificaciones importantes del proceso de producción incluyendo cualquier cambio en el equipo, condiciones de las áreas de producción o en los materiales que puedan influir en la calidad del producto o en la reproducibilidad del proceso.
16.7 De la revalidación.
Deben establecerse procesos y procedimientos sobre la base de un estudio de validación y deben ser objeto de revalidación periódica de forma crítica para garantizar que siguen siendo capaces de proporcionar los resultados previstos.
17. QUEJAS, RECLAMOS Y RETIRO DE PRODUCTOS.
17 .1 Generalidades
17.1.1 De las generalidades.
Todo reclamo, queja, retiro o cualquier información relativa a productos posiblemente defectuosos deben ser objeto de una investigación de acuerdo a procedimientos escritos. Debe existir un sistema para retirar del mercado en forma rápida y efectiva un producto cuando éste tenga un defecto o exista sospecha de ello.
17 .2 Reclamos.
17.2.1 De la persona responsable del seguimiento de la queja o reclamo.
Los procedimientos deben indicar la persona responsable de atender las quejas y reclamos, y decidir qué medidas deben adoptarse en conjunto con personal de otros departamentos involucrados que la asistan en esta tarea.
17.2.2 De los procedimientos de quejas y reclamos.
El fabricante debe contar con procedimientos escritos para el manejo de productos devueltos por quejas o reclamos, que debe incluir, como mínimo, lo siguiente:
a. Nombre del producto.
b. Forma y presentación farmacéutica.
c. Código o número de lote del producto
d. Fecha de expiración.
e. Nombre y datos generales de la persona que realizó el reclamo.
f. Fecha del reclamo.
g. Motivo de reclamo.
h. Revisión de las condiciones del producto cuando se recibe.
i. Investigación que se realiza.
j. Determinación de las acciones correctivas y medidas adoptadas.
17.2.3 De la investigación en otros lotes
Si se descubre o sospecha un defecto en un lote deben evaluarse otros lotes que pudieran haber sido afectados.
17.2.4 Del registro de acciones y medidas adoptadas
Se debe tomar nota de todas las acciones y medidas adoptadas como resultado de una queja y referirlas a los registros correspondientes al lote en cuestión.
17.2.5 De la revisión periódica
Los registros de reclamos deberán revisarse periódicamente para buscar cualquier indicación de problemas específicos o repetitivos que requieran acción especial y el eventual retiro de productos comercializados.
17.2.6 De la información a las autoridades competentes
Se deberá informar a la Autoridad Reguladora cuando un fabricante considere necesario tomar alguna medida en relación con una fabricación defectuosa, deterioro o cualquier otro problema grave de calidad de un producto.
17.3 Retiros
17.3.1 De la orden
La orden de retiro de un producto del mercado, debe ser emitida por la Autoridad Reguladora o por el fabricante.
17.3.2 Del responsable
Debe asignarse un responsable de la coordinación independiente del departamento de ventas, y la ejecución del retiro de productos se debe realizar de acuerdo a un procedimiento escrito.
17.3.3 De los procedimientos
Debe haber procedimientos establecidos por escrito, comprobados y actualizados periódicamente, con el fin de organizar las actividades de retiro en forma ágil y rápida. Este debe incluir la notificación inmediata a las autoridades correspondientes de los diferentes países.
17.3.4 Del acceso
El responsable del retiro debe tener fácil acceso a los registros de distribución
17.3.5 Del informe
Debe registrarse el proceso de retiro y redactarse un informe sobre el mismo, como también conciliarse los datos relacionados con las cantidades de producto distribuido y retirado.
17.3.6 De la identificación y almacenamiento de productos retirados
Los productos retirados se identificarán y almacenarán independientemente en un área segura mientras se esté a la espera de una decisión sobre su destino final.
18 AUTO INSPECCIÓN Y AUDITORIAS DE CALIDAD
18.1 Auto inspección
18.1.1 De las generalidades
El laboratorio fabricante debe realizar auto inspecciones y auditorias periódicas.
18.1.2 Del procedimiento y programa de auto inspección
Se debe definir un procedimiento y un programa de auto inspección para verificar el cumplimiento de las Buenas Prácticas de Manufactura y emitir un informe que incluya las evaluaciones, resultados, conclusiones y las medidas correctivas recomendadas.
18.1.3 Del seguimiento
Todas las recomendaciones, referentes a medidas correctivas, deben ponerse en práctica. El procedimiento de auto inspección debe documentarse y debe instituirse un programa efectivo de seguimiento y cumplimiento de las acciones correctivas
18.1.4 De la frecuencia
La auto inspección debe efectuarse en forma regular por lo menos una vez al año. Se podrán hacer en forma parcial, garantizando que, a lo largo de un año, todos los departamentos del laboratorio fabricante han sido auditados
18.1.5 Del personal que ejecuta la auto inspección
El personal asignado para realizar la auto inspección debe ser un grupo de expertos en sus respectivos campos y tener conocimiento de las Buenas Prácticas de Manufactura con el fin de evaluar de forma objetiva todos los sistemas.
18.1.6 De la guía para realizar auto inspección
Para realizar auto inspecciones se podrá utilizar la "Guía de Verificación de Buenas Prácticas de Manufactura para la Industria Farmacéutica" armonizada por los países centroamericanos.
18.2 Auditorías
18.2.1 Generalidades
Como complemento de las auto inspecciones se deben realizar auditorías de calidad, las cuales deben extenderse a los proveedores y contratistas locales y a los externos cuando las leyes de cada país lo permitan.
18.2.2 Del personal que ejecuta la auditoría
La auditoría de la calidad puede ser realizada por expertos independientes al Laboratorio fabricante o bien por un equipo designado por la administración del laboratorio, específicamente con ese fin.
18.2.3 Del procedimiento
Se debe definir un procedimiento para la realización de auditorías para detectar cualquier deficiencia en la garantía de calidad y emitir un informe que incluya los resultados y conclusiones.
18.2.4 De las Auditorías o inspección del Autoridad Reguladora
Todo laboratorio fabricante queda sujeto a auditorias o inspecciones periódicas por parte de la Autoridad Reguladora.
19. VIGILANCIA Y VERIFICACIÓN
La vigilancia y verificación del cumplimiento del presente Reglamento le corresponde a la autoridad reguladora de cada país centroamericano. En casos justificados, las autoridades sanitarias podrán verificar el cumplimiento de las buenas prácticas de manufactura de los laboratorios fabricantes establecidos fuera de los países centroamericanos, aplicando el presente reglamento técnico.
19.1 De las Auditorías o Inspección de la Autoridad Reguladora.
19.1.1 Las inspecciones para la verificación del cumplimiento del presente reglamento se harán utilizando la "Guía de verificación de buenas prácticas de manufactura para la industria farmacéutica"
20 ELEMENTOS COMPLEMENTARIOS
20.1 Concordancia y Correspondencia
-Informe 32 Series de informes técnicos 8 23, Comité de expertos de la OMS en especificaciones para las preparaciones Farmacéuticas Ginebra 1992
-Reglamento de Buenas Prácticas de Manufactura de la Industria Farmacéutica, Anexo 3 de la resolución 93-2002 (COMIECO XXIV)
21 Bibliografía
Arias, Tomas D Glosario de Medicamentos: desarrollo, evaluación y uso.
Organización Panamericana de la Salud.
Washington, D.C.: OPS, c 1999, 333p
Reglamento de Buenas Prácticas de Manufactura en las Plantas Farmacéuticas. Ministerio de Salud, Costa Rica, Agosto 2003.
Reglamento de Buenas Prácticas de Manufactura para Laboratorios de Productos Farmacéuticos de Uso Humano. Ministerio de Salud Pública y Asistencia Social. Departamento de Regulación y Control de Productos Farmacéuticos y Afines. Guatemala, Marzo 2004.
Normas de Correcta Fabricación. Volumen 4. Normas sobre Medicamentos de la Unión Europea. Comisión Europea. Edición 1999.
Anexo A
(Normativo)
A. Fabricación de Productos Farmacéuticos Estériles.
A. 1 Generalidades
A. 1.1 De los requisitos
El Laboratorio fabricante de estos productos deben tener permiso o licencia sanitaria de funcionamiento correspondiente y cumplir con el Reglamento de Buenas Prácticas de Manufactura, en lo que aplique, además de lo indicado en este anexo.
A. 1. 2 De los requisitos especiales.
La producción de productos farmacéuticos estériles debe estar sujeta a requisitos especiales para minimizar los riesgos de contaminación microbiana, de partículas y de pirógenos
A. 1.3 De las áreas de producción.
La producción de productos farmacéuticos estériles debe realizarse en áreas limpias en las que el ingreso debe ser a través de esclusas para el personal, equipos y materiales.
A. 1.4 De las operaciones.
Las operaciones de preparación de materiales, producción y esterilización deben llevarse a cabo en áreas separadas dentro del área limpia.
A. 1.5 De las categorías de producción. Las operaciones de producción de estériles se deben realizar por alguna de las siguientes categorías
A) Producción aséptica
B) Producción con esterilización final
C) Producción con esterilización por filtración
A. 1.6 De las Condiciones.
El diseño de las áreas debe garantizar que se alcancen los niveles de calidad del aire en funcionamiento o en reposo
A 1.7 De la clasificación de las áreas.
Las áreas limpias destinadas a la fabricación de preparaciones estériles deben de cumplir con la clasificación en grados A, B, C, D.
NOTA N°. 1
Grado A
Es el área específica de operaciones de alto riesgo. Estas condiciones se consiguen normalmente en cabina de flujo laminar. Los sistemas de flujo laminar deben proporcionar una velocidad homogénea del aire de 0.45 mis+/ - 20% en el punto de trabajo.
Grado B
Entorno para el área de grado A, en el caso de preparación y llenado aséptico.
Grado C y D
Áreas limpias para realizar fases menos críticas de la fabricación de medicamentos estériles.
TABLA A 1-CLASIFICACIÓN DE PARTÍCULAS DE AIRE CORRESPONDIENTE A LOS
DIFERENTES GRADOS
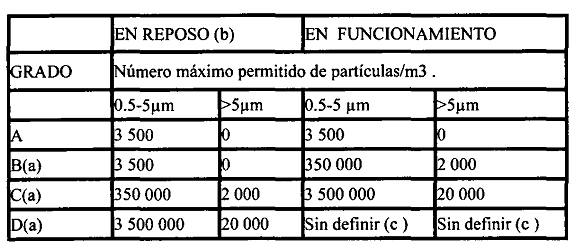
(a) Con el fin de alcanzar los grados de aire B, C, D, el número de renovaciones del aire debe ser proporcional al tamaño del área, del equipo y personal presente en la misma. El sistema de aire debe estar provisto de filtros HEPA, para los grados A, B y C, los cuales deberán estar ubicados al nivel del techo o pared.
(b) Las orientaciones dadas para el número máximo permitido de partículas en la situación "en reposo", corresponde aproximadamente a la Norma Federal 209 E de Estados Unidos de Norteamérica y a las clasificaciones ISO de la forma siguiente: Los grados A y B se corresponde con la clase 100, el grado C con la clase 10,000 y el grado D con la clase 100,000.
(c) El requisito y límite de ésta área de penderá de la naturaleza de las operaciones que se realicen en ella.
TABLA A-2 OPERACIONES QUE DEBEN REALIZARSE EN LOS DIVERSOS GRADOS.
A 1.8 Del control de partículas
A fin de controlar el nivel de partículas de los distintos grados se debe monitorear las áreas" en funcionamiento" y documentarlo
A. 1.9 Del monitoreo microbiológico de las áreas.
Se deben realizar controles microbiológicos de las distintas áreas en funcionamiento y documentarlos. Los resultados deben incluirse en la documentación del lote para su evaluación al momento de la liberación del producto terminado.
NOTA No. 2
(a) Las placas de sedimentación individuales deben exponerse no menos de cuatro horas.
A 1.10 De los límites de alerta.
Debe establecerse límites de alerta, si estos se superan se deben adoptar medidas correctivas según los resultados del monitoreo.
A 2 1 Producción Aséptica
A. 2.1 Generalidades
La producción aséptica se debe realizar con materiales estériles y en ambiente grado A con entorno de grado B.
A. 2.2 De la elaboración y llenado
La elaboración y llenado de formas farmacéuticas líquidas, sólidas y semisólidas estériles deben realizarse en un ambiente de grado A con un entorno de grado B, cuando el producto esté expuesto.
A. 2.3 Del traslado de recipientes parcialmente cerrados.
El traslado de recipientes parcialmente cerrados, como los utilizados en la liofilización, antes de completar su cerrado, debe realizarse en un ambiente de grado A con un entorno de grado B o bien en bandejas de transporte cerradas en un ambiente de grado B.
A 3 Producción con esterilización final.
A. 3.1 Generalidades.
Las soluciones deben elaborarse en un ambiente de grado C, con el objeto de obtener conteos microbianos y particulados bajos, aptos para filtración y esterilización inmediatas. Las soluciones pueden elaborarse en ambiente de grado D, siempre que se tomen medidas adicionales para reducir al mínimo la contaminación, con el uso de reactores herméticos.
El llenado de preparaciones parenterales, debe efectuarse en un área de trabajo con flujo de aire laminar Grado A, en un ambiente de grado C y las no parenterales se llenarán en un ambiente grado C. La elaboración y el llenado de productos estériles semisólidos deben realizarse en un ambiente de grado Cantes de la esterilización final.
A. 3.2 De las condiciones de riesgo.
Cuando exista un riesgo inusual de contaminación para el producto por el entorno, debido a que la operación de llenado sea lenta, los recipientes tengan cuello ancho o estén expuestos necesariamente durante más de unos segundos antes de su cierre, el llenado debe hacerse en un área de grado A con un entorno al menos de grado C.
A. 4 Producción con esterilización por filtración
A. 4.1 Generalidades.
La manipulación de la materia prima y la preparación de soluciones deben realizarse en un ambiente grado C. Estas actividades pueden efectuarse también un ambiente grado D, siempre que se tomen medidas adicionales para reducir al mínimo la contaminación, con el uso de reactores herméticos antes de la filtración. Luego de la filtración estéril, el producto debe manipularse y llenarse en recipientes bajo condiciones estériles en un área de grado A o B con ambiente de grado B o C respectivamente.
A. 5 Personal.
A. 5.1 Del número de personas en áreas limpias
En las áreas limpias sólo debe estar presente el número mínimo de personas necesarias; esto es especialmente importante durante la producción aséptica. Las inspecciones y controles deben realizarse desde afuera de las áreas limpias.
A. 5.2 De la capacitación específica del personal El personal
(incluyendo el personal de limpieza y mantenimiento), que trabajan en dichas áreas deben someterse regularmente a programas de capacitación relacionadas con la Buenas prácticas de manufactura de productos estériles, incluyendo la higiene y conocimientos básicos de microbiología. El ingreso de personas extrañas a las áreas estériles, que no hayan recibido dicha capacitación, deben ser supervisadas cuidadosamente.
A. 5.3 De las condiciones de higiene y salud del personal
El personal involucrado en la fabricación de preparaciones estériles debe mantener altos niveles de higiene y limpieza. Tienen la obligación· de informar sobre cualquier situación de salud que pueda influir en las condiciones ambientales. Deben efectuarse exámenes periódicos al personal para determinar si existen dichas condiciones.
A. 5.4 Del ingreso a las áreas limpias.
A las áreas limpias no deben ingresar personas que visten ropa de calle y el personal que ingresa a los vestidores debe usar uniforme o vestimenta de uso en el laboratorio. Con respecto al cambio, lavado de ropa y aseo personal se deben seguir procedimientos escritos.
A. 5.5 De las restricciones
Las personas que ingresan en las áreas limpias no deben usar reloj de pulsera, joyas, celulares, radio localizador, cosméticos y ningún instrumento ajeno al uniforme de los cuales puedan desprenderse partículas.
A. 5.6 Del uniforme o vestimenta.
El tipo y material de ropa debe ser de acuerdo al proceso de producción y al grado del área; el uniforme o vestimenta debe usarse de tal forma que los productos estén protegidos de la contaminación.
NOTA No. 3 Del tipo de uniforme o vestimenta de acuerdo al grado del área
Uniforme o vestimenta necesaria para cada grado de área:
a. Grado D.
Debe quedar cubierto totalmente el cabello, la barba y bigote.
Debe usar un uniforme o vestimenta y zapatos o cubre-zapatos.
b. Grado C
Debe quedar cubierto totalmente el cabello, la barba y el bigote.
Debe llevarse un uniforme o vestimenta de una o dos piezas, ajustado en las muñecas, con cuello alto, con zapatos o cubre zapatos. Esta ropa no debe liberar ninguna fibra ni partícula.
c. Grado A/ B
Un cobertor de cabeza o escafandra que debe cubrirla totalmente; los bordes inferiores de dicho cobertor deben introducirse dentro del cuello del traje; debe llevarse un uniforme o vestimenta de una o dos piezas, ajustado en las muñecas, con cuello alto. Debe usarse mascarilla, guantes estériles de látex que no contengan talco, calzado esterilizado o desinfectado; la parte inferior de los pantalones deben introducirse dentro del calzado y los extremos de las mangas del uniforme deben introducirse dentro de los guantes. El uniforme o vestimenta no debe liberar ninguna fibra ni partícula y debe retener las partículas producidas por el cuerpo.
A. 5.7 De las precauciones del uso de uniforme o vestimenta
El uniforme del área no estéril no debe introducirse al vestidor que conduce directamente a las áreas de grado B y C. El personal de las áreas de grado A, B o C debe usar uniforme limpio y estéril, según procedimiento escrito. El uniforme, las mascarillas y los guantes se deben cambiar en cada sesión de trabajo.
A. 5.8 Del lavado de uniformes o vestimentas.
La limpieza y el lavado del uniforme utilizado en el área limpia deben efectuarse de tal forma que no se adhieran partículas contaminantes que posteriormente puedan desprenderse de las mismas. Se debe disponer dentro de las instalaciones de lavandería con un área dedicada para este tipo de uniformes. Las operaciones de lavado deben efectuarse de conformidad con procedimientos.
A. 5.9 De la esterilización de uniformes.
Las operaciones de esterilización de uniformes o vestimentas deben efectuarse de conformidad con procedimientos.
A. 6 Instalaciones.
A. 6.1 De diseño de las instalaciones.
Las instalaciones deben estar diseñadas para reducir el ingreso de personal de supervisión o control y debe permitir que todas las operaciones puedan ser observadas desde el exterior.
A. 6.2 De las características de las áreas.
En las áreas limpias, todas las superficies expuestas deben ser lisas, impermeables, sin fisuras, con curvas sanitarias, que permitan la aplicación repetida de agentes de limpieza, y sanitización. No se permiten puertas corredizas.
A. 6.3 De los cielos rasos o cielos falsos.
Los cielos falsos o cielos rasos, deben ser lisos y sellarse herméticamente para prevenir la contaminación proveniente del espacio libre.
A. 6.4 De las tuberías, duetos y otros servicios.
Las tuberías, duetos y otros servicios deben estar empotrados e instalados de manera que faciliten su limpieza, para evitar contaminación o acumulación de polvo.
A. 6.5 De las instalaciones de lavado.
Las instalaciones para el lavado de utensilios y drenajes son prohibidas en las áreas de grado A, B o C utilizadas para la fabricación aséptica. Cuando sea necesario instalarlas en las áreas grado D, debe diseñarse, ubicarse y mantenerse de tal manera que se reduzca al mínimo el riesgo de contaminación microbiana. Deben contar con drenajes y tapas tipo sanitario.
A. 6.6 De los vestidores.
Los vestidores deben estar diseñados como esclusas y se utilizarán para proporcionar separación física de las diferentes etapas de cambio de uniformes, para minimizar así la contaminación microbiana y por partículas del uniforme o vestimenta. Los vestidores deben contar con diferenciales de presión. La fase final del vestidor debe tener en situación de reposo, el mismo grado que el área a la que conduzca. Las instalaciones para el lavado de manos deben estar ubicadas solamente en la primera fase de los vestidores, nunca en los lugares donde se realizan trabajos asépticos.
A. 6.7 De las esclusas.
Se debe contar con esclusas para el acceso de personas y materiales, las cuales deben diseñarse para reducir la contaminación procedente del entorno
A. 6.8 Del paso a través de esclusas.
Las puertas de una esclusa no deben abrirse simultáneamente. Debe disponerse de un sistema de cierre ínter bloqueado o un sistema de alarma visual / auditivo para prevenir la apertura simultánea de las puertas.
A. 6.9 De la clasificación del aire en las esclusas.
La clasificación del aire requerida para la esclusa depende de su diseño y aplicación. Debe controlarse y en caso de elaboración aséptica, debe ser al menos de grado D.
A. 6.10 De la efectividad de las esclusas
Las esclusas deben utilizarse sólo después de verificar su efectividad. Esta verificación debe tener en cuenta todos los factores críticos de la tecnología de las esclusas, como la calidad del aire del interior y del exterior (entorno) de la esclusa, sanitización, proceso de transferencia e integridad de la esclusa.
A. 7 Sistemas de aire
A. 7.1 De los diferenciales de presión.
La entrada de aire filtrado debe mantener una presión positiva y un flujo de aire respecto a las áreas adyacentes de grado menor en todas las condiciones de trabajo y debe barrer eficazmente al área. Las áreas adyacentes de grados diferentes deben tener un gradiente de presión.
A. 7.2 De las medidas de control
Debe instalarse un sistema de alarma para detectar las fallas en el suministro de aire. Las áreas entre las cuales es importante que haya diferenciales de presión deben disponer del indicador correspondiente.
Los diferenciales de presión se registrarán periódicamente quedando documentados.
A. 7.3 Del mantenimiento.
Cuando sea posible las operaciones de mantenimiento y reparaciones deben realizarse fuera del área estéril, de lo contrario debe llevarse a cabo de acuerdo a un procedimiento que garantice la esterilidad del área o proceso.
A. 8 Sistemas de agua
A. 8.1 Del agua para productos farmacéuticos estériles
El agua para productos farmacéuticos estériles se producirá, conservará, distribuirá y almacenará de manera que se evite el crecimiento microbiano, de acuerdo con procedimientos establecidos.
A. 8.2 De la obtención del agua.
La obtención de agua para productos farmacéuticos estériles debe ser a partir de agua previamente tratada por diferentes mecanismos de purificación.
A. 8.3 Del muestreo.
Debe existir un procedimiento de muestreo en el cual se definan los puntos y su rotación.
A. 8.4 Del monitoreo.
El agua para la producción de productos farmacéuticos estériles debe monitorearse periódicamente en diferentes puntos de muestreo para detectar contaminación química, microbiológica y endotoxinas. Debe conservarse registros de los resultados del monitoreo y de cualquier medida adoptada.
A. 8.5 De la conservación.
En la producción, almacenamiento y distribución de agua, se debe impedir el crecimiento microbiano, recurriendo a una circulación constante con una temperatura de 80 ºC o no más de 4 ºC, cuando se requiera almacenar para ser utilizada en la producción.
A. 8.6 Del uso.
Cada lote de producción de agua para inyectable debe ser aprobado por Control de Calidad previa realización de los controles analíticos físico-químicos, microbiológicos y endotoxinas bacterianas, excepto cuando existan sistemas validados.
A. 9 Equipo
A .9.1 De la esterilización.
Los equipos que se utilicen en los procesos de fabricación de productos estériles, deben ser esterilizados por medio de vapor, calor seco u otros métodos.
A. 9.2 Del mantenimiento.
Siempre que sea posible los equipos, accesorios y servicios deben diseñarse e instalarse de forma que las operaciones, el mantenimiento y las reparaciones se realicen fuera del área limpia. El equipo reparado cuando ingresa a un área limpia se sanitizará y esterilizarán las partes del equipo, cuando sea posible.
A. 9.3 Del mantenimiento dentro del área.
Cuando el mantenimiento de los equipos se efectúa dentro del área limpia, deben emplearse instrumentos y herramientas esterilizados, y el área debe ser sanitizada antes de iniciar el proceso.
A. 9.4 Del mantenimiento preventivo.
Todo el equipo, los sistemas de esterilización, sistema de aire, sistemas de tratamiento y almacenamiento de agua, deben ser objeto de mantenimiento planificado; y su posterior utilización deberá ser aprobada.
A. 10 Sanitización.
A. 10.1 Del área La sanitización del área limpia es especialmente importante.
Debe limpiarse de acuerdo a un procedimiento y a un programa de rotación de los sanitizantes Y controlarse periódicamente
A. 10. 2 De los sanitizantes y detergentes
Los sanitizantes y detergentes que se utilicen, deben someterse a control en cuanto a su contaminación microbiana; las diluciones se deben mantener en recipientes limpios e identificados conservándose durante un periodo definido. Si un recipiente está parcialmente vacío no debe completarse a volumen.
A. 10.3 Del monitoreo microbiológico de las áreas
Durante las operaciones, las áreas limpias deben monitorearse mediante el conteo microbiano de aire y superficie a intervalos programados. Cuando se llevan a cabo operaciones asépticas, dicho monitoreo debe ser más frecuente para asegurar que el ambiente esté dentro de las especificaciones. Debe tenerse en cuenta los resultados del monitoreo, para la posterior autorización de los lotes. Se debe controlar también regularmente la calidad de aire respecto a la cantidad de partículas. Después de la validación de sistemas, sanitización o limpieza es conveniente efectuar controles, aun cuando no se efectúen operaciones de producción.
A .11 Producción
A. 11.1 Generalidades.
Durante todo el proceso de producción deben tomarse precauciones para minimizar la contaminación, incluidas las fases previas a la esterilización.
A. 11.2 De la actividad.
Las actividades efectuadas en las áreas limpias deben reducirse al mínimo, especialmente cuando se están efectuando operaciones asépticas, y el movimiento de personal debe ser controlado y metódico, para evitar la liberación excesiva de partículas y microorganismos. La temperatura y humedad del ambiente deben controlarse teniendo en cuenta la naturaleza de la vestimenta utilizada.
A. 11.3 De la presencia de envases y materiales.
Debe reducirse al mínimo la presencia de envases y materiales que puedan desprender fibras en áreas limpias y evitarse completamente cuando se está efectuando un proceso aséptico.
A. 11.4 Del manejo e identificación de componentes, envases y equipos.
Los componentes, envases y equipos deben manipularse de forma que no se contaminen después de su sanitización. Deben identificarse adecuadamente de acuerdo a la etapa del proceso.
A. 11.5 Del tiempo de utilización de componentes, envases y equipo.
El intervalo entre el lavado, secado y esterilización de los componentes, recipientes de productos a granel y equipos, como también el intervalo entre la esterilización y el uso, deben ser lo más breves posibles y deben someterse a un límite de tiempo según procedimiento escrito.
A. 11.6 Del intervalo de preparación y filtración.
Debe definirse un tiempo máximo autorizado entre el inicio de la preparación de una solución y su esterilización o filtración a través de un filtro de retención microbiana, para cada producto, teniendo en cuenta su composición y el método de almacenamiento previsto.
A. 11.7 De los límites de contaminación.
La contaminación microbiológica debe ser mínima antes de la esterilización. Todas las soluciones, especialmente las destinadas a parenterales de gran volumen, deben pasar a través de un filtro de esterilización inmediatamente antes del proceso de llenado. Cuando se trata de soluciones acuosas en recipientes cerrados herméticamente deben estar protegidos todos los orificios de salida de presión.
A. 11.8 Del ingreso de materiales, equipo.
El ingreso de los materiales, envases y equipos en el área limpia, cuando se esté realizando un trabajo aséptico, deben esterilizarse e introducirse en el área mediante equipos de esterilización de doble puerta situados en la pared, o mediante un procedimiento que proporcione el mismo resultado de no introducir contaminantes.
A. 11.9 Del suministro de gases.
Los gases no combustibles deben suministrarse filtrado a través de filtros de retención microbiana (filtros esterilizantes).
A. 11.10 De la comprobación de las operaciones asépticas
El empleo de medios de cultivo que estimulan el crecimiento bacteriano en ensayos destinados a simular las operaciones asépticas (llenado de medios estériles, "llenado de caldos"), constituye un factor importante de la comprobación general de un proceso aséptico.
Nota No. 4
Tales ensayos deben reunir las siguientes características:
a. Debe simular lo más fielmente posible operaciones reales teniendo en cuenta factores tales como: complejidad de las operaciones, número de empleados que están trabajando y el tiempo de duración.
b. Debe ser posible que en el (los) medio(s) seleccionado(s) se pueda cultivar un amplio espectro de microorganismos incluyendo aquellos que se esperaría encontrar en un ambiente donde se efectúa el llenado.
c. Debe incluir número suficiente de unidades de producción para que se tenga alto grado de seguridad de que, al existir, podrían ser detectados aún niveles bajos de contaminación.
Se recomienda la inclusión de un mínimo de 3,000 unidades de producción en cada llenado de caldo. Se debe procurar llegar al nivel 0 de crecimiento debiendo ser considerada inaceptable cualquier cifra posterior a 0.1% de unidades contaminadas. Toda contaminación debe ser investigada. Los llenados de caldo deben repetirse a intervalos regulares, y siempre que tenga que efectuarse una comprobación como resultado de alguna alteración significativa en la producción, instalaciones, equipos u operaciones de procesado.
A. 12 Esterilización
A. 12.1 Generalidades.
Se puede efectuar la esterilización por medio de: calor húmedo o seco, óxido de etileno (u otro agente esterilizador apropiado), por filtración o por radiación ionizante. Cada método tiene sus aplicaciones y limitaciones particulares.
A. 12.2 De la validación de los procesos de esterilización.
Deben validarse y documentarse todos los procesos de esterilización.
A. 12.3 De la validez del proceso.
Debe demostrarse que el proceso de esterilización es eficaz para alcanzar los niveles de esterilización deseados, según procedimiento escrito. La validez del proceso deberá verificarse a intervalos programados, como mínimo una vez al año, y siempre que se ha introducido modificaciones significativas al equipo. Deben conservarse registros de los resultados.
A. 12.4 De los indicadores biológicos.
En caso que se utilicen indicadores biológicos, deben adoptarse precauciones estrictas para evitar la transferencia de contaminación microbiana a partir de los mismos. Deben almacenarse y utilizarse de acuerdo con las instrucciones del fabricante y su calidad se comprobará mediante controles positivos
A. 12.5 De la identificación de las cargas.
Debe existir un procedimiento escrito para evitar la confusión de los productos que han sido esterilizados de aquellos que no han sido esterilizados.
Nota No. 5
Pueden utilizarse indicadores como cinta de autoclave, cuando sea apropiado, para indicar si un lote (o sublote) ha pasado o no por un proceso de esterilización, pero estos indicadores no aseguran de forma confiable que el lote sea estéril en realidad.
A. 12.6 De los registros Deben existir registros de cada ciclo de esterilización.
Estos registros se verificarán como parte del procedimiento de aprobación del lote.
A. 13 Esterilización por calor
A. 13.1 Del control y registro de temperatura.
Cada ciclo de esterilización por calor debe registrarse mediante equipo apropiado, y con la debida precisión. La temperatura debe registrarse en el punto más frío de la carga o de la cámara cargada habiéndose determinado éste punto durante la validación; ésta debe ser verificada. Los controles deben formar parte del registro del lote. Pueden emplearse indicadores químicos o biológicos, pero éstos no deben reemplazar a los controles efectuados por medios físicos.
A. 13.2 Del tiempo.
Debe dejarse tiempo suficiente para que toda la carga alcance la temperatura necesaria antes de iniciar el cómputo de tiempo de esterilización. Dicho tiempo tendrá que determinarse para cada tipo de carga que se vaya a tratar.
A. 14 Esterilización por calor húmedo.
A. 14.1 Del control y registro de temperatura y presión.
La esterilización por calor húmedo es apropiada solamente para materiales que pueden humedecerse y para soluciones acuosas. Para controlar éste proceso debe tenerse en cuenta la temperatura y la presión. Normalmente el instrumento que registra la temperatura debe ser independiente del utilizado para el control y se debe utilizar un indicador de temperatura también independiente, cuya lectura de temperatura debe comprobarse durante el período de esterilización. Si se trata de esterilizadores que tienen drenaje en el fondo de la cámara es necesario registrar también la temperatura en esa posición durante todo el período de esterilización. Debe comprobarse frecuentemente la ausencia de fugas en la cámara cuando forme parte del ciclo una fase de vacío.
A. 14.2 De los materiales a esterilizar.
Los materiales a esterilizar, que no estén en envases cerrados, deben empacarse en material que permita la eliminación del aire y la penetración del vapor pero que impida la contaminación después de la esterilización. Todas las partes de la carga deben estar en contacto con el agente esterilizador a la temperatura requerida durante el tiempo necesario.
A. 14.3 Del vapor.
Debe asegurarse que el vapor utilizado en la esterilización tiene la calidad necesaria y que no contenga aditivos en un grado que pudiera provocar la contaminación del producto o del equipo.
A. 15 Esterilización por calor seco.
A. 15.1 Del proceso de esterilización por calor seco.
En el proceso de esterilización con calor seco, el aire debe circular dentro de la cámara, manteniéndose una presión positiva para impedir la entrada de aire no estéril. El aire suministrado debe pasar a través de un filtro HEPA. Cuando este proceso tenga también el objetivo de eliminar los pirógenos, debe utilizarse como parte de la validación pruebas con carga de endotoxinas.
A. 16 Esterilización por radiación.
A. 16.1 Del proceso de esterilización por radiación.
La esterilización por radiación se utiliza principalmente para esterilizar materiales y productos sensibles al calor.
Nota No. 6:
Muchos medicamentos y algunos materiales de acondicionamiento son sensibles a las radiaciones, por lo que éste métodos ó lo podrá permitirse cuando se haya confirmado experimentalmente la ausencia de efectos nocivos sobre el producto.
A. 16.2 De la dosis de radiación
Durante el proceso de esterilización debe medirse la dosis de radiación empleando dosímetros independientes de la tasa de radiación, que indique una medida cuantitativa de la dosis recibida por el producto mismo. Los dosímetros deben insertarse en la carga en número adecuado y suficientemente cercano entre ellos para asegurar que haya un dosímetro en la cámara en todo momento. Cuando se trata de dosímetros plásticos deben emplearse dentro del tiempo límite según su calibración. Debe verificarse la absorbancia del dosímetro poco después de su exposición a la radiación. Los indicadores biológicos pueden emplearse solamente como un control adicional. Los discos de colores sensibles a la radiación pueden usarse para distinguir entre los envases sometidos a la radiación y aquellos que no; dichos discos no son indicadores de una esterilización adecuada. La información obtenida debe formar parte del registro del lote.
A. 16.3 De los envases.
Los procedimientos de validación deben garantizar que se tienen en cuenta los efectos de las variaciones en la densidad de los envases.
A. 16.4 De la identificación de las cargas
Los procedimientos de manipulación de materiales deben evitar la confusión entre materiales irradiados y no irradiados. Cada paquete debe llevar discos de color sensibles a la radiación para distinguir entre envases que se ha sometido a la radiación y los que no.
A. 16.5 Del tiempo de radiación
La dosis de radiación total debe administrarse durante un periodo de tiempo determinado previamente.
A. 17 Esterilización con óxido de etileno.
A. 17 .1 Del proceso de esterilización.
Este método solo debe utilizarse cuando no pueda aplicarse ningún otro. Durante la validación del proceso, debe demostrarse que no se produce ningún efecto nocivo sobre el producto y que las condiciones y el tiempo permitidos para la liberación del gas son suficientes para reducir el gas residual y los productos de reacción a límites aceptables definidos según el tipo de producto o material.
A. 17.2 De las precauciones.
Es fundamental el contacto directo entre el gas y las células microbianas; deben tomarse precauciones para evitar la presencia de microorganismos que puedan estar encerrados en materiales como cristales o proteínas desecadas. La naturaleza y la cantidad de los materiales de acondicionamiento pueden afectar al proceso de forma significativa.
A. 17.3 De las condiciones.
Antes de su exposición al gas, debe establecerse un equilibrio entre los materiales, la humedad y temperatura requerida por el proceso. El tiempo empleado en esta operación debe considerarse en relación con la necesidad de reducir al mínimo posible el tiempo transcurrido antes de la esterilización
A. 17.4 De los indicadores biológicos
Cada ciclo de esterilización debe controlarse con indicadores biológicos apropiados, utilizando el número de unidades de indicadores de acuerdo al tamaño de la carga y distribuidas por toda la carga. La información así obtenida deberá incluirse en la documentación del lote.
A. 17.5 Del manejo de los indicadores biológicos
Los indicadores biológicos deben ser almacenados y utilizados de conformidad con las instrucciones del fabricante y su desempeño debe ser verificado mediante controles positivos.
A. 17.6 Del control y registro.
En cada ciclo de esterilización se llevarán registros del tiempo empleado en completar el ciclo, de la presión, temperatura, humedad dentro de la cámara durante el proceso, y de la concentración del gas, así como de la cantidad total de gas utilizada. La presión y la temperatura deben registrarse a lo largo de todo el ciclo en una gráfica. Los registros deben incluirse en la documentación del lote.
A. 17.7 De la eliminación del gas residual
Después de la esterilización, la carga debe conservarse de forma controlada en condiciones de ventilación que permitan que el gas residual y los productos de reacción se reduzcan hasta el nivel definido. Este proceso debe ser validado.
A. 18 Filtración de productos que no pueden esterilizarse en su envase final.
A. 18.1 Del proceso de filtración
Los productos que no pueden ser esterilizados en el recipiente final, pueden ser filtrados a través de un filtro estéril con poros de 0.22 micras (o menos), o de uno con características equivalentes de retención de microorganismos, en recipientes previamente esterilizados.
A. 18.2 Del uso de pre filtro o doble filtración.
Debido a los posibles riesgos potenciales del método de filtración respecto a otros procesos de esterilización, puede realizarse una filtración utilizando pre filtros y filtros de retención microbiana o dos filtraciones en donde la segunda filtración se realiza inmediatamente antes del llenado utilizando filtros de retención microbiana. La filtración estéril final debe realizarse lo más cerca posible al punto de llenado.
A. 18.3 De los filtros.
No debe emplearse filtros que desprendan fibras, ni que contengan asbesto.
A. 18.4 De la integridad del filtro.
La integridad del filtro esterilizado debe comprobarse antes de su utilización e inmediatamente después de su utilización por un método aprobado, como la prueba de punto de burbuja, flujo de difusión o mantenimiento de la presión. El tiempo empleado en filtrar un volumen conocido de solución a granel y la diferencia de presión que debe aplicarse en el filtro debe determinarse durante la validación y será necesario registrar e investigar cualquier diferencia importante que se de en estos parámetros durante la fabricación normal. Los resultados de estos controles deben quedar registrados en la documentación del lote.
A. 18.5 Del uso del filtro.
No debe utilizarse el mismo filtro durante más de un día de trabajo a menos que las especificaciones del filtro permitan la reutilización previa esterilización, según procedimiento escrito.
18.6 De la composición de los filtros.
El filtro no debe afectar al producto, reteniendo componentes de éste, ni añadiéndole sustancias.
A. 19 Acabado de productos estériles.
A. 19.1 De sellado o cerrado de envases.
El cierre y sellado de los envases debe ser efectuado mediante métodos debidamente comprobados, verificando la integridad por medio de muestras representativas según procedimiento escrito.
A. 19.2 De los envases cerrados al vacío.
En los envases cerrados al vacío se comprobará el mantenimiento de este vacío tras un periodo previamente determinado.
A. 19.3 De la inspección visual.
Los envases de productos parenterales llenos deben inspeccionarse al 100 por ciento. Si la inspección es visual, debe efectuarse bajo condiciones controladas de iluminación y fondo. Si se utilizan otros métodos de inspección, éstos también deben validarse y los aparatos empleados deben ser controlados a intervalos regulares. Los resultados quedarán registrados
Nota No. 7
Los inspectores deben someterse a controles regulares de vista con anteojos puestos, si los usan normalmente y durante las inspecciones deben tener descansos frecuentes.
A. 20 Control de Calidad.
A. 20.1 De la prueba de esterilidad.
La prueba de esterilidad a la que se somete el producto terminado debe ser considerada sólo como la última de una serie de medidas de control, mediante la cual se garantiza la esterilidad y sólo puede interpretarse como parte de un conjunto, que incluya los registros de las condiciones ambientales y el proceso de los lotes.
A. 20.2 De las muestras
Las muestras que se tomen para el ensayo de esterilidad deben ser representativas del conjunto del lote o lo que recomiendan las farmacopeas oficiales.
Nota No. 8
Entre ellas deben incluirse especialmente muestras tomadas de las partes del lote que se consideren con mayor riesgo de contaminación como:
a. En el caso de productos que se hayan llenado asépticamente, las muestras incluirán envases llenados al principio y al final del lote y después de cualquier interrupción significativa del trabajo.
b. En el caso de productos que se hayan sometido a esterilización por calor en su envase final, debe procurarse tomar muestras procedentes de la parte potencialmente más fría de la carga.
A. 20.3 De la aprobación de los lotes.
Los lotes que no cumplen la prueba inicial de esterilidad no pueden ser aprobados sobre la base de una segunda prueba, a menos que se lleve a cabo una investigación del tipo de organismo encontrado y de los registros sobre las condiciones ambientales y el procesado de los lotes, y como resultado de las misma se demuestre que la prueba original no era válida.
A. 20.4 Del monitoreo.
Cuando se trata de productos inyectables se debe monitorear el control del agua, los productos intermedios y productos terminados para verificar si contienen endotoxinas, empleando métodos establecidos por farmacopea o que hayan sido validados en casos de no ser oficiales, para cada tipo de producto. Para parenterales de gran volumen el control del agua o de los productos intermedios debe efectuarse en todos los casos, además de las pruebas del producto terminado. Cuando una muestra no pasa la prueba debe investigarse la causa y adoptarse las medidas correctivas necesarias.
ANEXO B
(NORMATIVO)
B. Fabricación de Productos Farmacéuticos â-lactámicos (Derivados penicilínicos y cefalosporinas)
B. 1 De los requisitos.
El Laboratorio fabricante debe tener Permiso sanitario de funcionamiento o licencia sanitaria correspondiente, cumplir con el Reglamento de Buenas Prácticas de Manufactura y el anexo de estériles cuando aplique; además de lo indicado en este anexo
B. 2 PERSONAL
B. 2.1 Del personal.
El laboratorio fabricante debe disponer de personal exclusivo para la fabricación de â-lactámicos y en caso de rotación, se debe pasar por un periodo de cuarentena no menor de siete días a menos que se cuente con un procedimiento validado para disminuir ese periodo.
B. 2.2 De la prueba de sensibilidad.
A todo el personal que labora en el laboratorio fabricante, se le debe realizar la prueba de sensibilidad para este tipo de producto, como mínimo una vez al año. A las personas autorizadas que ingresen al mismo se les debe realizar esta prueba previamente.
B. 2.3 De la capacitación.
El personal que labora en estas áreas debe recibir capacitación específica, y la efectividad práctica debe evaluarse en forma periódica.
B. 2.4 De la salida de personas.
El personal y toda persona autorizada que ingrese a las instalaciones, antes de salir del laboratorio fabricante, deben ducharse con agua y jabón alcalino, con el objeto de minimizar la contaminación.
B. 2.5 De los uniformes.
Los uniformes que usa el personal que está en contacto con los productos debe cubrir en su totalidad el cuerpo y ser de uso exclusivo. Antes de lavarse o desecharse deben descontaminarse o desactivarse, de acuerdo a un procedimiento escrito.
B. 2.6 Del equipo de protección
Los operarios deben usar equipo de protección durante todo el proceso productivo.
B. 3 Instalaciones
B. 3.1 Generalidades. Debe contar con edificios separados y autónomos o instalaciones independientes y autónomas para la fabricación de â-lactámicos.
B. 3.2 Del acceso.
El acceso a las áreas de producción debe ser solo para personas autorizadas, previa capacitación y de acuerdo a un procedimiento escrito
B. 3.3 De las esclusas.
Debe existir esclusas independientes para el ingreso de operarios y materiales, para todas las áreas de producción (a excepción de empaque secundario) dichas esclusas deben contar con diferenciales de presión que eviten la salida de contaminantes a las áreas adyacentes.
B. 3.4 De la sanitización de las áreas. Se debe contar con un procedimiento escrito para la desactivación y sanitización de las áreas.
B. 3.5 De las trazas en las áreas.
Se debe realizar análisis de trazas después de la desactivación y sanitización de las áreas, dejando registros de los mismos.
B. 3.6 Tratamiento de aguas residuales. Los laboratorios fabricantes deben de contar con un sistema para el tratamiento de aguas residuales, cumpliendo con los parámetros ambientales, establecidos en la legislación ambiental de cada país centroamericano.
B. 4 Sistema de aire
B. 4.1 De las características. El sistema de aire debe garantizar que el aire recirculado carece de contaminación y la fracción que sale al exterior esté libre de producto y contaminantes, utilizando filtros HEPA terminales.
B. 4.2 De los diferenciales de presión. Deben existir dispositivos para medir los diferenciales de presión y su correspondiente registro.
B. 4.3 De la desactivación, limpieza y destrucción Debe existir un procedimiento escrito y registro para la desactivación, limpieza de duetos, destrucción de residuos y filtros usados en las instalaciones.
B. 5 Equipos.
B. 5.1 Generalidades
Deben existir equipos exclusivos para estás áreas.
B. 5.2 De la sanitización
Se debe contar con un procedimiento escrito para la desactivación y sanitización del equipo.
B. 5.3 De las trazas.
Se debe realizar análisis de trazas después de la desactivación y sanitización de los equipos, dejando registros de estos.
B. 5.4 Del mantenimiento.
El mantenimiento preventivo de los equipos debe realizarse de acuerdo a un programa y procedimiento escrito, el cual se debe ejecutar dentro de las instalaciones, dejando registros.
B. 6 Control de calidad
B .6.1 Del traslado de las muestras para control de calidad.
Previo al traslado de las muestras para verificación de la calidad a otras instalaciones de la misma empresa se debe desactivar el recipiente de acuerdo a un procedimiento escrito.
ANEXO C
(NORMATIVO)
C. Fabricación de Productos con Hormonas y Productos Citostáticos.
C. 1 De los requisitos.
El Laboratorio fabricante de estos productos deben tener Permiso sanitario de funcionamiento o Licencia sanitaria correspondiente y cumplir con el Reglamento de Buenas Prácticas de Manufactura y el anexo de estériles cuando aplique; además de lo indicado en este anexo.
C. 2 Generalidades.
Para la fabricación de productos citostáticos se debe realizar en edificios separados y autónomos o instalaciones independientes y autónomas. En el caso de la fabricación de productos con hormonas se debe realizar en áreas segregadas, pudiéndose trabajar por campaña y validando sus procesos de limpieza y producción.
C. 3 Personal.
C. 3.1 De la protección.
Se debe proporcionar indumentarias protectoras con las siguientes características:
a) Uniformes protectores desechables confeccionados con materiales de baja permeabilidad para la producción de éstos productos.
b) En el caso de producción de productos estériles, la tela del uniforme no debe desprender partículas.
c) El uniforme debe ser manga larga, puños y tobillos ajustados.
d) Guantes de látex desechables, libre de talco.
e) Mascarilla o respirador de vapores y partículas con filtros HEPA.
f) Lentes protectores.
g) Cofia y escafandra
C. 3.2 De la capacitación.
El personal que labora en estas áreas debe recibir capacitación específica, y la efectividad práctica debe evaluarse en forma periódica.
C. 3.3 De la salida de personas. El personal y toda persona autorizada que ingrese a las instalaciones, deben ducharse antes de salir del laboratorio fabricante.
C. 3.4 De los controles clínicos
A todo el personal que labora en estas áreas, se le debe determinar los niveles hormonales y citostáticos, de acuerdo a procedimientos escritos.
C. 3.5 Del acceso del personal.
El acceso a las áreas de producción debe ser solo para personas autorizadas, previa capacitación y de acuerdo a un procedimiento escrito.
C. 4 Instalaciones
C. 4.1 De las esclusas. Deben existir esclusas independientes para el ingreso de operarios y materiales, para todas las áreas de producción (a excepción de empaque secundario), dichas esclusas deben contar con diferenciales de presión que impidan la salida de contaminantes a las áreas adyacentes.
C. 4.2 Sanitización de las áreas.
Debe de contar con un procedimiento escrito para la desactivación y sanitización de las áreas.
C. 4.3 De las trazas en las áreas
Se debe realizar análisis de trazas después de la desactivación y sanitización de las áreas, dejando registro de los mismos.
C. 4.4 Tratamiento de aguas residuales.
Los laboratorios fabricantes deben de contar con un sistema para el tratamiento de aguas residuales, cumpliendo con los parámetros ambientales, establecidos en la legislación ambiental de cada país centroamericano.
C. 5 Sistema de aire.
C. 5.1 De las características.
El sistema de aire utilizando filtro HEPA terminales, debe garantizar que el aire recirculado carece de contaminación y la fracción que sale al exterior esté libre de producto fabricado.
C. 5.2 De los diferenciales de presión.
Deben existir dispositivos para medir los diferenciales de presión y su correspondiente registro.
C. 5.3 De la desactivación, limpieza y destrucción.
Debe existir un procedimiento escrito y registro para la desactivación, limpieza de duetos, destrucción de residuos y filtros usados en las instalaciones.
C. 6 Equipos
C. 6.1 Generalidades.
Deben existir equipos exclusivos para estás áreas.
C. 6.2 De la sanitización.
Se debe contar con un procedimiento escrito para la desactivación y sanitización del equipo.
C. 6.3 De las trazas.
Se debe realizar análisis de trazas después de la desactivación y sanitización de los equipos, dejando registro de los mismos.
C. 6.4 Del mantenimiento.
El mantenimiento preventivo de los equipos debe realizarse de acuerdo a un programa y procedimiento escrito, el cual se debe ejecutar dentro de las instalaciones, dejando registros del mismo.
C. 6.5 De la eliminación de los residuos y materiales.
Debe existir un procedimiento escrito que contemple la inactivación e incineración de todos los residuos y materiales de limpieza, así como la indumentaria protectora desechable.
C. 7 Control de calidad
C. 7.1 Del traslado de las muestras para control de calidad
Previo al traslado de la muestra para verificación de la calidad a otras instalaciones de la misma empresa se debe desactivar el recipiente de acuerdo a un procedimiento escrito.
-Fin del Reglamento Técnico-
-------------------------------------------------------------------------------------------------------------------------
GUÍA DE VERIFICACIÓN DEL REGLAMENTO TÉCNICO CENTROAMERICANO RTCA 11.03.42:07 REGLAMENTO TÉCNICO SOBRE BUENAS PRÁCTICAS DE MANUFACTURA PARA LA INDUSTRIA FARMACÉUTICA. PRODUCTOS FARMACÉUTICOS Y MEDICAMENTOS DE USO HUMANO
Publicado en La Gaceta, Diario Oficial N°. 217 del 14 de noviembre de 2014
ANEXO DE LA RESOLUCIÓN No. 339-2014
(COMIECO-LXVII)
GUÍA DE VERIFICACIÓN DEL REGLAMENTO TÉCNICO CENTROAMERICANO RTCA 11.03.42:07 REGLAMENTO TÉCNICO SOBRE BUENAS PRÁCTICAS DE MANUFACTURA PARA LA INDUSTRIA FARMACÉUTICA. PRODUCTOS FARMACÉUTICOS Y MEDICAMENTOS DE USO HUMANO.
Editada por:
• Ministerio de Economía, MINECO
•Organismo Salvadoreño de Reglamentación Técnica, OSARTEC
• Ministerio de Fomento, Industria y Comercio, MIFIC
• Secretaría de Industria y Comercio, SIC
• Ministerio de Economía. Industria y Comercio, MElC
l. INTRODUCCIÓN
El RTCA I 1.03.42:07 Reglamento Técnico sobre Buenas Prácticas de Manufactura para la Industria Farmacéutica. Productos Farmacéuticos y Medicamentos de Uso Humano, establece que la verificación de su cumplimiento le corresponde a la Autoridad Reguladora de cada Estado Parte, lo que implica la revisión de todos los elementos relacionados con las BPM implementados en la industria, destinados a garantizar la producción de lotes uniformes de productos farmacéuticos con el* fin de asegurar la calidad, seguridad y eficacia de los mismos.
El presente documento consiste en el instrumento oficial para verificar el cumplimiento de las BPM en la industria farmacéutica, por parte de la Autoridad Reguladora de cada Estado Parte, con el cual se pretende homologar y armonizar los criterios de inspección y establecer una lista de puntos a verificar de todas las operaciones y procesos de la industria. Puede ser también de utilidad para los laboratorios fabricantes en lo que respecta al auto inspección.
Cada ítem tiene asignada una calificación con la finalidad de que las inspecciones a realizar, respondan a criterios uniformes de evaluación, dichos criterios se definen en el glosario del presente documento.
II. OBJETIVO
Establecer los criterios de evaluación a seguir por parte de la Autoridad Reguladora, para verificar el cumplimiento del RTCA 11.03.42:07 Reglamento Técnico sobre Buenas Prácticas de Manufactura para la Industria Farmacéutica. Productos Farmacéuticos y Medicamentos de Uso Humano.
III. ALCANCE
Esta guía es de aplicación a todos los laboratorios farmacéuticos establecidos en el territorio de los Estados Parte.
IV. DOCUMENTOS A CONSULTAR
El RTCA 11.03.42:07 Reglamento Técnico sobre Buenas Prácticas de Manufactura para la Industria Farmacéutica. Productos Farmacéuticos y Medicamentos de Uso Humano.
V. RESPONSABLE
Autoridad Reguladora de cada Estado Parte.
VI. GLOSARIO
CRITERIO CRÍTICO: aquel que en atención a las recomendaciones de las Buenas Prácticas de Manufactura, afecta en forma grave e inadmisible la calidad, seguridad de los productos y la seguridad de los trabajadores, en su interacción con los productos y procesos.
CRITERIO MAYOR: aquel que en atención a las recomendaciones de las Buenas Prácticas de Manufactura, puede afectar en forma grave la calidad, seguridad de los productos y seguridad de los trabajadores, en su interacción con los productos y procesos.
CRITERIO MENOR: aquel que en atención a las recomendaciones de las Buenas Prácticas de Manufactura, puede afectar en forma leve la calidad, seguridad de los productos y seguridad de los trabajadores, en su interacción con los productos y procesos.
Observación La Resolución N°. 339-2014 (COMIECO-LXVII), fue publicada en La Gaceta, Diario Oficial N°. 209 del 04 de noviembre de 2014, y su anexo a esta resolución en La Gaceta, Diario Oficial N°. 217 del 14 de noviembre de 2014.